- Филогенетический анализ и молекулярная эволюция. Лекция 5

Содержание
- 2. Задачи молекулярной эволюции Изучение законов изменения наследственной информации в живых системах, включая неклеточные и клеточные формы
- 3. Филогенетический анализ и молекулярная эволюция На молекулярном уровне эволюция является процессом мутации и селекции Молекулярная эволюция
- 4. Квагга (вымерла) больше похожа на зебру или лошадь?
- 5. 23.10.2019 Кафедра биоинформатики МБФ РНИМУ
- 6. Мутации Ошибки, происходящие при репликации генома Могут происходить как в половых, так и в соматических клетках
- 7. Классификации мутаций По числу затронутых нуклеотидов (по длине мутации) Точечные мутации (point mutations) Изменения в нескольких
- 8. Классификации мутаций По типам событий, происходящих при мутации Замена одного нуклеотида на другой (nucleotide substitution) Вставка
- 9. В кодирующих участках вставки и делеции могут приводить к сдвигу рамки считывания => изменение всей аминокислотной
- 10. Нуклеотидные замены Транзиции Замена пурина на другой пурин: A ? G или G ? A Замена
- 11. Нуклеотидные замены Трансверсии Замена между пуринами и пиримидинами: A ? T A ? C G ?
- 12. SNP Однонуклеотидный полиморфизм (англ. Single nucleotide polymorphism, SNP) — любая единичная замена основания (A, T, G
- 13. 23.10.2019 Кафедра биоинформатики МБФ РНИМУ
- 14. Нонсенс и миссенс мутации Замена кодирующего триплета на стоп-кодон – нонсенс мутация (nonsence mutation) Замена кодирующего
- 15. Эволюция нуклеотидной последовательности 11111111112222222222233 12345678901234567890123456789012 0 ATCTATACGGTCGATGCTAGCTGATCGATCGA 1 -------------------------------- 2 ------T--------A---------------- 3 ---------------C------C--------- 4 -----...-----T-C------CG-------- 5
- 16. Эволюция нуклеотидной последовательности 11111111112222222222233 12345678901234567890123456789012 0 ATCTATACGGTCGATGCTAGCTGATCGATCGA 1 -------------------------------- 2 ------T--------A---------------- 3 ---------------C------C--------- 4 -----...-----T-C------CG-------- 5
- 17. Эволюция нуклеотидной последовательности Идентичные Похожие Находятся на определенной эволюционной дистанции Первичные и вторичные замены Обратная замена
- 18. Эволюция нуклеотидной последовательности Дивергенция – разделение на независимые родственные эволюционные линии Параллельные мутации – мутации, произошедшие
- 19. Эволюция нуклеотидной последовательности Гомологичные последовательности – последовательности, имеющие общее эволюционное происхождение Группа гомологичных последовательностей, произошедших от
- 20. 23.10.2019 Кафедра биоинформатики МБФ РНИМУ
- 21. Эволюция нуклеотидной последовательности MRCA – most recent common ancestor (наиболее недавний общий предок) LUCA – last
- 22. Консенсусные последовательности Искусственная последовательность, содержащая в каждой позиции нуклеотид, встречаемый наиболее часто у анализируемых последовательностей Обычно,
- 23. Консенсусные последовательности В консенсусной последовательности можно отображать разнообразие нуклеотидов в конкретной позиции (гетерогенность позиции) Если в
- 24. 23.10.2019 Кафедра биоинформатики МБФ РНИМУ
- 25. Концепция молекулярных часов Закрепление мутаций в популяции занимает определённое время и постоянна Цукеркандль и Поллинг сформулировали
- 26. Концепция молекулярных часов Если известно, что дивергенция двух, различающихся между собой на один нуклеотид последовательностей произошла
- 27. 23.10.2019 Кафедра биоинформатики МБФ РНИМУ
- 28. Влияние отбора на мутации Мутации, улучшающие приспособленность организма, подвергаются действию положительного естественного отбора — эволюционным силам,
- 29. Измерение отбора путем анализа последовательностей белок-кодирующих генов (Hurst, 2002; Li, 1997) Отношение Ka/Ks (где Ka –
- 30. Ka/Ks = 1 – нейтральная эволюция белковой последовательности (кодируемый белок не подвергается отбору). Для большинства белок-кодирующих
- 31. Использование Ka/Ks для измерения уровня отбора предполагает нейтральность синонимичных сайтов. Однако Ka и Ks положительно коррелируют
- 32. Критерий Макдональда – Крейтмана (Aquadro, 1997; McDonald and Kreitman, 1991) широко используется для измерения отбора. Он
- 33. Филогенетические деревья 23.10.2019 Кафедра биоинформатики МБФ РНИМУ The time will come, I believe, though I shall
- 34. Причина подобия – общее происхождение! 23.10.2019 Кафедра биоинформатики МБФ РНИМУ
- 35. Гомология – происхождение от общего предка Подобие – наблюдаемые данные, собранные сейчас не подразумевающие каких-либо исторических
- 36. Зачем нужны филогенетические деревья? Биологические задачи: сравнение 3-х и более объектов (кто на кого более похож
- 37. Реальные события : Данные: Построенное дерево эволюция в природе или в например, древовидный граф, лаборатории, а.к.
- 38. Основные термины Узел (node) — точка разделения предковой последовательности (вида, популяции) на две независимо эволюционирующие. Соответствует
- 39. Какие бывают деревья? Бинарное (разрешённое) (в один момент времени может произойти только одно событие ) Небинарное
- 40. Какие бывают деревья? Укорененное дерево (rooted tree) отражает направление эволюции Неукорененное (бескорневое) дерево (unrooted tree) показывает
- 41. Рутинная процедура, или как строят деревья? Составление выборки последовательностей Множественное выравнивание Построение дерева фрагмент записи в
- 42. (((C:3.2,D:8.0):5.5,E:7.7):5.2,(A:6.1,B:6.3):7.5); длины ветвей (((C,D),E)),(A,B)); только топология Скобочная формула (Newick format) A B C D E 5.2
- 43. Как выбирать последовательности для дерева? Кроме случаев очень близких последовательностей, проще работать с белками (а не
- 44. Самое главное – хорошее выравнивание! Максимальный вклад в финальное дерево: нельзя построить хорошее дерево по плохому
- 45. Основные алгоритмы построения филогенетических деревьев Методы, основанные на оценке расстояний (матричные методы): Вычисляются эволюционные расстояния между
- 46. Пример матрицы расстояний 1 2 3 4 5 6 7 8 0.00 10.53 9.77 12.78 12.03
- 47. Как понимать расстояние между объектами? Как время, в течение которого они эволюционировали Как число «эволюционных событий»
- 48. Гипотеза «молекулярных часов» (E.Zuckerkandl, L.Pauling, 1962) За равное время во всех ветвях эволюции накапливается равное число
- 49. UPGMA Unweighted Pair Group Method with Arithmetic Mean разновидность кластерного метода Расстояние между кластерами вычисляется как
- 50. 23.10.2019 Кафедра биоинформатики МБФ РНИМУ
- 51. Гипотеза молекулярных часов не всегда справедлива A B C D E (длина ветвей пропорциональна числу мутаций)
- 52. Недостатки UPGMA Алгоритм строит ультраметрическое дерево, а это означает, что скорость эволюции предполагается одинаковой для всех
- 53. Метод ближайших соседей (Neighbor-joining, NJ) Строит неукоренённое дерево Может работать с большим количеством данных Достаточно быстрый
- 54. Метод Neighbor-joining Рисуем «звездное» дерево и будем «отщипывать» от него по паре листьев Пусть ui =
- 55. Метод ближайших соседей (Neighbor-joining, NJ) 2. Кластер (i, j) – новый узел дерева Расстояние от i
- 56. Стандартная ситуация Понимаем расстояние как число мутаций Реальное (неизвестное нам) дерево — укоренённое, но не ультраметрическое
- 57. Как изобразить дерево? Топология дерева Топология дерева — только листья, узлы, (корень) и связывающие их ветви
- 58. Филограмма: Длина ребер пропорциональна эволюционному расстоянию между узлами. Кладограмма: представлена только топология, длина ребер игнорируется. Arabidopsis
- 59. Достоверность топологии. Bootstraps Создадим псевдоданные: N множественных выравниваний той же длины, что и исходное, каждое из
- 60. Какие on-line программы строят деревья? ClustalW. “Tree type” – nj, phylip: строит только методом NJ, но
- 61. Phylip 23.10.2019 Кафедра биоинформатики МБФ РНИМУ
- 62. Пакет Phylip protdist — оценка эволюционных расстояний между белковыми последовательностями (вход — множественное выравнивание, выход —
- 63. Bootstrapping with Phylip Надо выбрать Bootstrap options в protdist, выставить не менее 100 итераций, нечетное число
- 64. Общий план действий с пакетом Phylip Множественное выравнивание -> protdist Bootstrap options - ? Результат –
- 66. Скачать презентацию

Задачи молекулярной эволюции
Изучение законов изменения наследственной информации в живых системах, включая
Задачи молекулярной эволюции
Изучение законов изменения наследственной информации в живых системах, включая

Филогенетический анализ и молекулярная эволюция
На молекулярном уровне эволюция является процессом мутации
Филогенетический анализ и молекулярная эволюция
На молекулярном уровне эволюция является процессом мутации

Квагга (вымерла) больше похожа на зебру или лошадь?
Квагга (вымерла) больше похожа на зебру или лошадь?
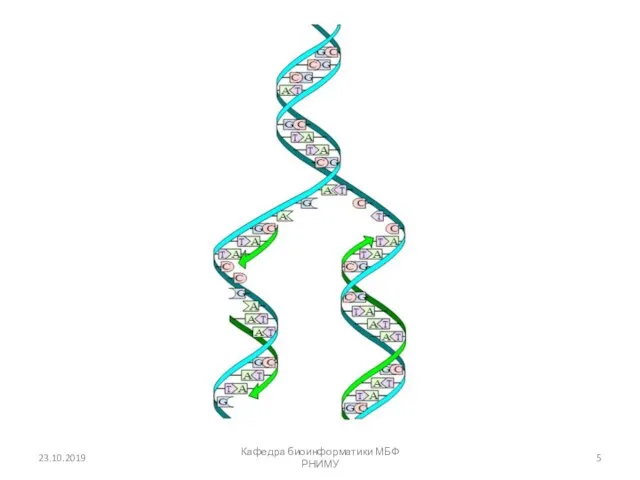
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
23.10.2019
Кафедра биоинформатики МБФ РНИМУ

Мутации
Ошибки, происходящие при репликации генома
Могут происходить как в половых, так и
Мутации
Ошибки, происходящие при репликации генома
Могут происходить как в половых, так и

Классификации мутаций
По числу затронутых нуклеотидов (по длине мутации)
Точечные мутации (point mutations)
Изменения
Классификации мутаций
По числу затронутых нуклеотидов (по длине мутации)
Точечные мутации (point mutations)
Изменения

Классификации мутаций
По типам событий, происходящих при мутации
Замена одного нуклеотида на
Классификации мутаций
По типам событий, происходящих при мутации
Замена одного нуклеотида на

В кодирующих участках вставки и делеции могут приводить к сдвигу рамки
В кодирующих участках вставки и делеции могут приводить к сдвигу рамки

Нуклеотидные замены
Транзиции
Замена пурина на другой пурин:
A ? G или G ?
Нуклеотидные замены
Транзиции
Замена пурина на другой пурин:
A ? G или G ?

Нуклеотидные замены
Трансверсии
Замена между пуринами и пиримидинами:
A ? T
A ? C
G ?
Нуклеотидные замены
Трансверсии
Замена между пуринами и пиримидинами:
A ? T
A ? C
G ?

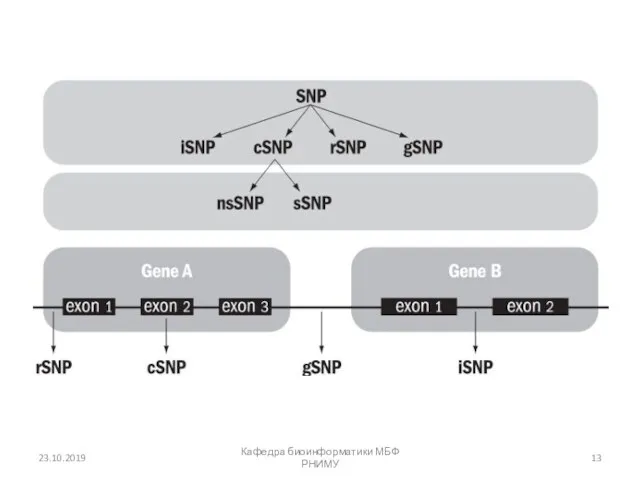
SNP
Однонуклеотидный полиморфизм (англ. Single nucleotide polymorphism, SNP) — любая единичная замена
SNP
Однонуклеотидный полиморфизм (англ. Single nucleotide polymorphism, SNP) — любая единичная замена
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
23.10.2019
Кафедра биоинформатики МБФ РНИМУ

Нонсенс и миссенс мутации
Замена кодирующего триплета на
стоп-кодон – нонсенс мутация
Нонсенс и миссенс мутации
Замена кодирующего триплета на стоп-кодон – нонсенс мутация

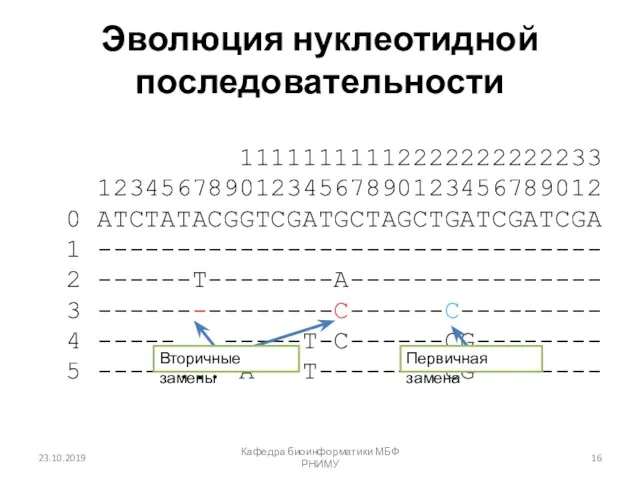
Эволюция нуклеотидной последовательности
11111111112222222222233
12345678901234567890123456789012
0 ATCTATACGGTCGATGCTAGCTGATCGATCGA
1 --------------------------------
2 ------T--------A----------------
3 ---------------C------C---------
4 -----...-----T-C------CG--------
5
Эволюция нуклеотидной последовательности
11111111112222222222233
12345678901234567890123456789012
0 ATCTATACGGTCGATGCTAGCTGATCGATCGA
1 --------------------------------
2 ------T--------A----------------
3 ---------------C------C---------
4 -----...-----T-C------CG--------
5
Эволюция нуклеотидной последовательности
11111111112222222222233
12345678901234567890123456789012
0 ATCTATACGGTCGATGCTAGCTGATCGATCGA
1 --------------------------------
2 ------T--------A----------------
3 ---------------C------C---------
4 -----...-----T-C------CG--------
5
Эволюция нуклеотидной последовательности
11111111112222222222233
12345678901234567890123456789012
0 ATCTATACGGTCGATGCTAGCTGATCGATCGA
1 --------------------------------
2 ------T--------A----------------
3 ---------------C------C---------
4 -----...-----T-C------CG--------
5

Эволюция нуклеотидной последовательности
Идентичные
Похожие
Находятся на определенной эволюционной дистанции
Первичные и вторичные замены
Обратная замена
Эволюция нуклеотидной последовательности
Идентичные
Похожие
Находятся на определенной эволюционной дистанции
Первичные и вторичные замены
Обратная замена

Эволюция нуклеотидной последовательности
Дивергенция – разделение на независимые родственные эволюционные линии
Параллельные мутации
Эволюция нуклеотидной последовательности
Дивергенция – разделение на независимые родственные эволюционные линии
Параллельные мутации

Эволюция нуклеотидной последовательности
Гомологичные последовательности – последовательности, имеющие общее эволюционное происхождение
Группа гомологичных
Эволюция нуклеотидной последовательности
Гомологичные последовательности – последовательности, имеющие общее эволюционное происхождение
Группа гомологичных

23.10.2019
Кафедра биоинформатики МБФ РНИМУ
23.10.2019
Кафедра биоинформатики МБФ РНИМУ

Эволюция нуклеотидной последовательности
MRCA – most recent common ancestor (наиболее недавний общий
Эволюция нуклеотидной последовательности
MRCA – most recent common ancestor (наиболее недавний общий

Консенсусные последовательности
Искусственная последовательность, содержащая в каждой позиции нуклеотид, встречаемый наиболее часто
Консенсусные последовательности
Искусственная последовательность, содержащая в каждой позиции нуклеотид, встречаемый наиболее часто

Консенсусные последовательности
В консенсусной последовательности можно отображать разнообразие нуклеотидов в конкретной позиции
Консенсусные последовательности
В консенсусной последовательности можно отображать разнообразие нуклеотидов в конкретной позиции
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
23.10.2019
Кафедра биоинформатики МБФ РНИМУ

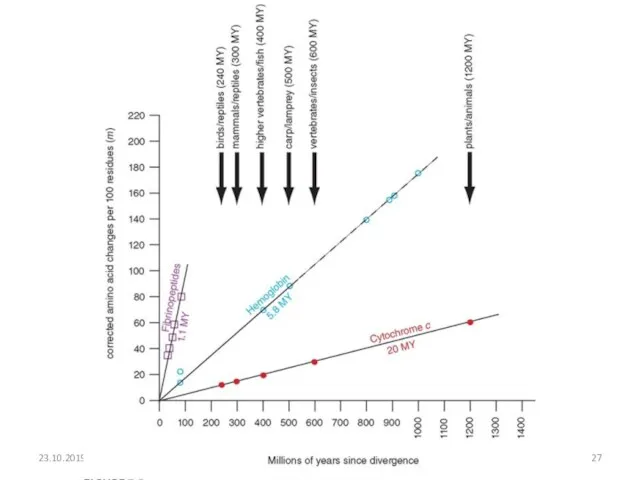
Концепция молекулярных часов
Закрепление мутаций в популяции занимает определённое время и постоянна
Цукеркандль
Концепция молекулярных часов
Закрепление мутаций в популяции занимает определённое время и постоянна
Цукеркандль

Концепция молекулярных часов
Если известно, что дивергенция двух, различающихся между собой на
Концепция молекулярных часов
Если известно, что дивергенция двух, различающихся между собой на
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
23.10.2019
Кафедра биоинформатики МБФ РНИМУ

Влияние отбора на мутации
Мутации, улучшающие приспособленность организма, подвергаются действию положительного естественного
Влияние отбора на мутации
Мутации, улучшающие приспособленность организма, подвергаются действию положительного естественного

Измерение отбора путем анализа последовательностей белок-кодирующих генов (Hurst, 2002; Li, 1997)
Отношение
Измерение отбора путем анализа последовательностей белок-кодирующих генов (Hurst, 2002; Li, 1997)
Отношение

Ka/Ks = 1 – нейтральная эволюция белковой последовательности (кодируемый белок не
Ka/Ks = 1 – нейтральная эволюция белковой последовательности (кодируемый белок не

Использование Ka/Ks для измерения уровня отбора предполагает нейтральность синонимичных сайтов.
Однако Ka
Использование Ka/Ks для измерения уровня отбора предполагает нейтральность синонимичных сайтов.
Однако Ka

Критерий Макдональда – Крейтмана (Aquadro, 1997; McDonald and Kreitman, 1991) широко используется
Критерий Макдональда – Крейтмана (Aquadro, 1997; McDonald and Kreitman, 1991) широко используется

Филогенетические деревья
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
The time will come, I believe, though
Филогенетические деревья
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
The time will come, I believe, though

Причина подобия – общее происхождение!
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
Причина подобия – общее происхождение!
23.10.2019
Кафедра биоинформатики МБФ РНИМУ

Гомология – происхождение от общего предка
Подобие – наблюдаемые данные, собранные сейчас
Гомология – происхождение от общего предка
Подобие – наблюдаемые данные, собранные сейчас

Зачем нужны филогенетические деревья?
Биологические задачи:
сравнение 3-х и более объектов
(кто
Зачем нужны филогенетические деревья?
Биологические задачи:
сравнение 3-х и более объектов
(кто
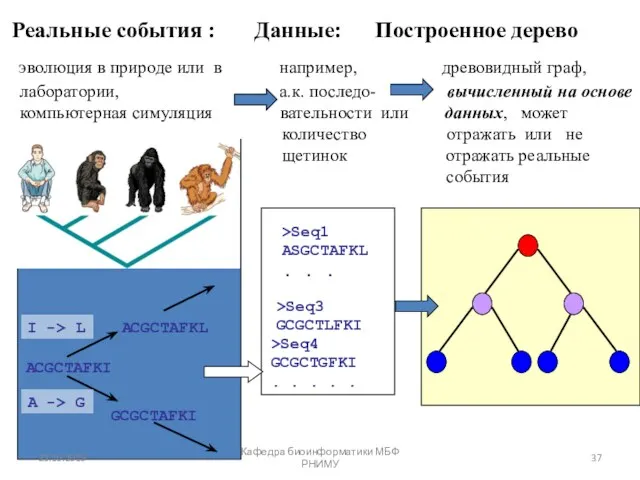
Реальные события : Данные: Построенное дерево
эволюция в природе
Реальные события : Данные: Построенное дерево
эволюция в природе
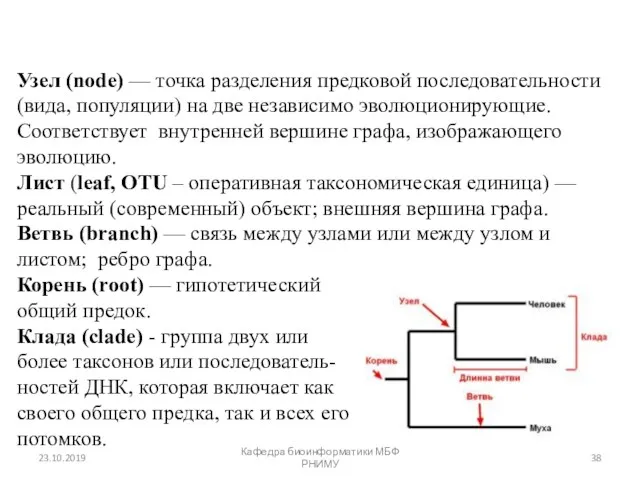
Основные термины
Узел (node) — точка разделения предковой последовательности
(вида, популяции) на две
Основные термины
Узел (node) — точка разделения предковой последовательности (вида, популяции) на две
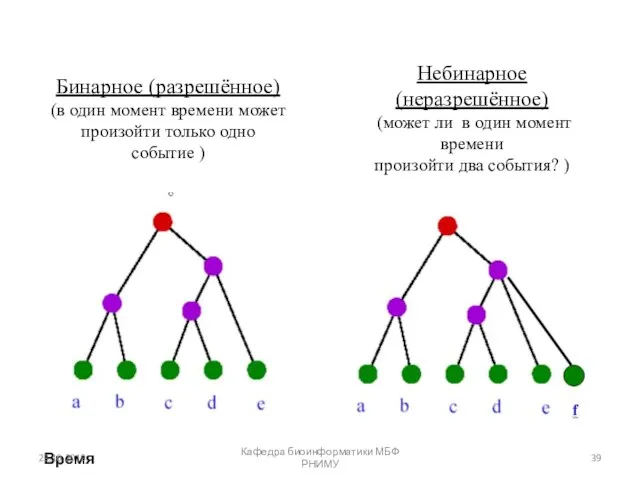
Какие бывают деревья?
Бинарное (разрешённое)
(в один момент времени может
произойти только одно
Какие бывают деревья?
Бинарное (разрешённое)
(в один момент времени может
произойти только одно
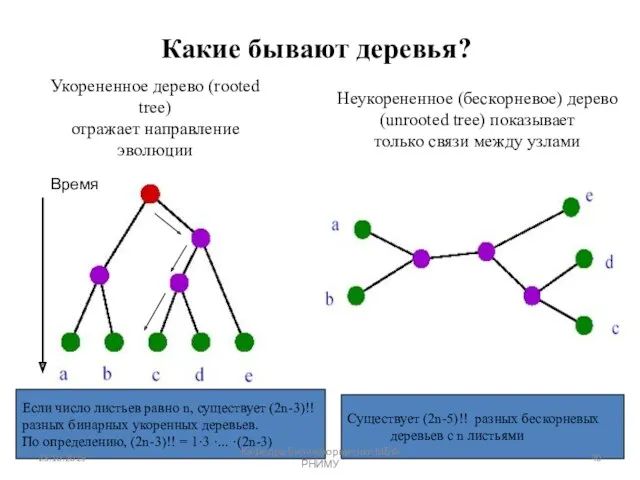
Какие бывают деревья?
Укорененное дерево (rooted tree)
отражает направление эволюции
Неукорененное (бескорневое) дерево
(unrooted tree)
Какие бывают деревья?
Укорененное дерево (rooted tree)
отражает направление эволюции
Неукорененное (бескорневое) дерево (unrooted tree)
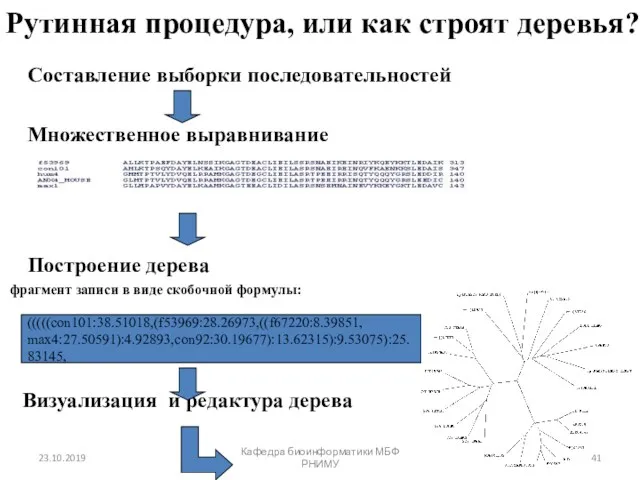
Рутинная процедура, или как строят деревья?
Составление выборки последовательностей
Множественное выравнивание
Построение
Рутинная процедура, или как строят деревья?
Составление выборки последовательностей
Множественное выравнивание
Построение

(((C:3.2,D:8.0):5.5,E:7.7):5.2,(A:6.1,B:6.3):7.5); длины ветвей
(((C,D),E)),(A,B)); только топология
Скобочная формула (Newick format)
A
B
C
D
E
5.2
7.5
6.3
6.1
7.7
8.0
3.2
5.5
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
(((C:3.2,D:8.0):5.5,E:7.7):5.2,(A:6.1,B:6.3):7.5); длины ветвей
(((C,D),E)),(A,B)); только топология
Скобочная формула (Newick format)
A
B
C
D
E
5.2
7.5
6.3
6.1
7.7
8.0
3.2
5.5
23.10.2019
Кафедра биоинформатики МБФ РНИМУ

Как выбирать последовательности для дерева?
Кроме случаев очень близких последовательностей, проще работать
Как выбирать последовательности для дерева?
Кроме случаев очень близких последовательностей, проще работать

Самое главное – хорошее выравнивание!
Максимальный вклад в финальное дерево: нельзя построить
Самое главное – хорошее выравнивание!
Максимальный вклад в финальное дерево: нельзя построить

Основные алгоритмы построения филогенетических деревьев
Методы, основанные на оценке
расстояний (матричные методы):
Вычисляются
Основные алгоритмы построения филогенетических деревьев
Методы, основанные на оценке
расстояний (матричные методы):
Вычисляются

Пример матрицы расстояний
1 2 3 4 5 6 7 8
Пример матрицы расстояний
1 2 3 4 5 6 7 8

Как понимать расстояние между объектами?
Как время, в течение которого они
Как понимать расстояние между объектами?
Как время, в течение которого они

Гипотеза «молекулярных часов»
(E.Zuckerkandl, L.Pauling, 1962)
За равное время во всех ветвях эволюции
Гипотеза «молекулярных часов»
(E.Zuckerkandl, L.Pauling, 1962)
За равное время во всех ветвях эволюции

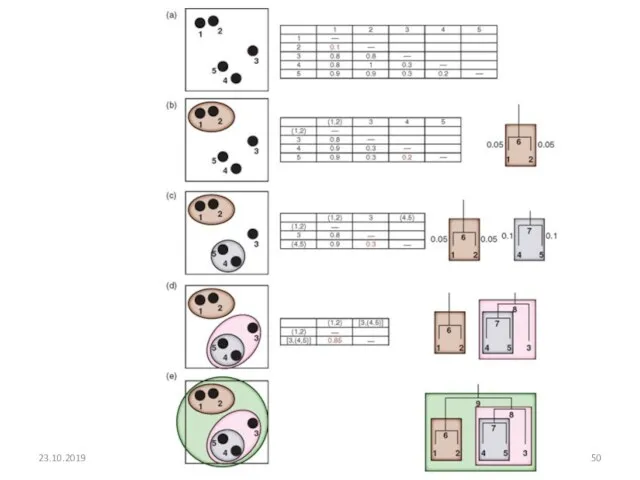
UPGMA
Unweighted Pair Group Method with Arithmetic Mean
разновидность кластерного метода
Расстояние между кластерами
UPGMA
Unweighted Pair Group Method with Arithmetic Mean
разновидность кластерного метода
Расстояние между кластерами
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
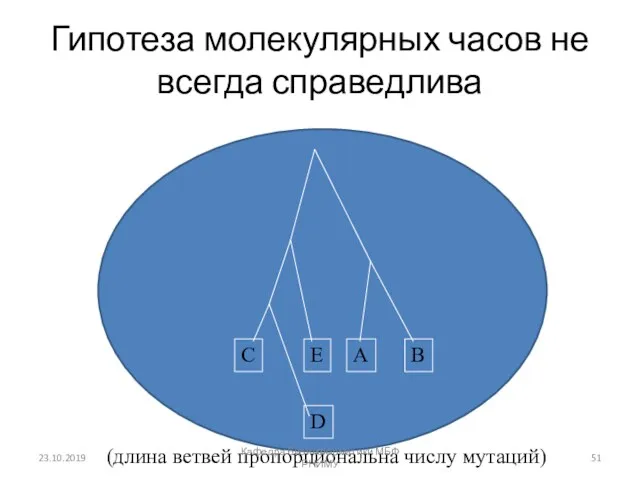
Гипотеза молекулярных часов не всегда справедлива
A
B
C
D
E
(длина ветвей пропорциональна числу мутаций)
23.10.2019
Кафедра биоинформатики
Гипотеза молекулярных часов не всегда справедлива
A
B
C
D
E
(длина ветвей пропорциональна числу мутаций)
23.10.2019
Кафедра биоинформатики
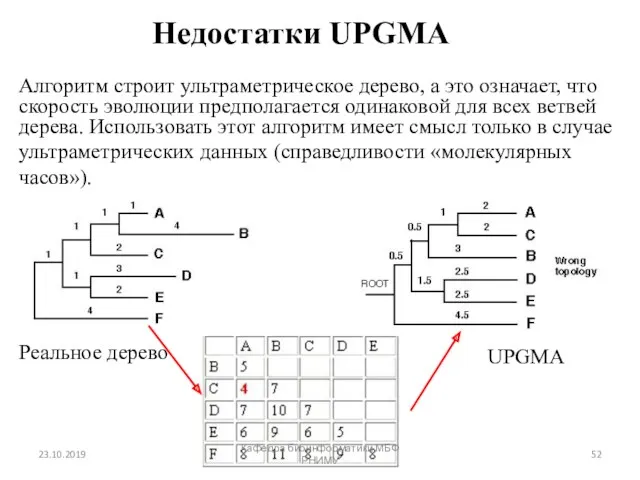
Недостатки UPGMA
Алгоритм строит ультраметрическое дерево, а это означает, что
скорость
Недостатки UPGMA
Алгоритм строит ультраметрическое дерево, а это означает, что
скорость

Метод ближайших соседей
(Neighbor-joining, NJ)
Строит неукоренённое дерево
Может работать с большим количеством
Метод ближайших соседей
(Neighbor-joining, NJ)
Строит неукоренённое дерево
Может работать с большим количеством

Метод Neighbor-joining
Рисуем «звездное» дерево и будем «отщипывать» от него по
Метод Neighbor-joining
Рисуем «звездное» дерево и будем «отщипывать» от него по
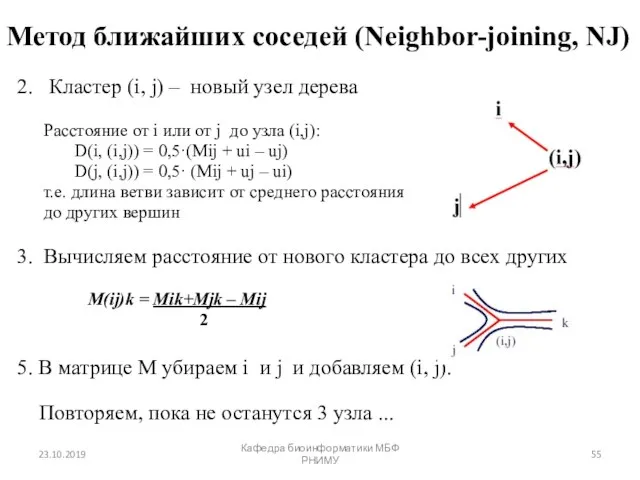
Метод ближайших соседей (Neighbor-joining, NJ)
2. Кластер (i, j) – новый узел
Метод ближайших соседей (Neighbor-joining, NJ)
2. Кластер (i, j) – новый узел

Стандартная ситуация
Понимаем расстояние как число мутаций
Реальное (неизвестное нам)
Стандартная ситуация
Понимаем расстояние как число мутаций
Реальное (неизвестное нам)
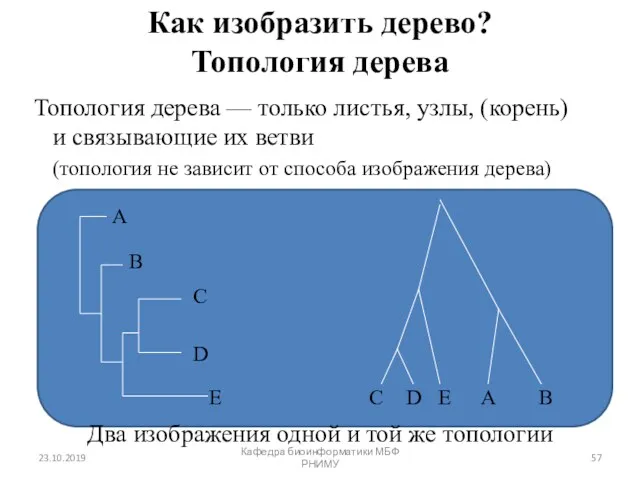
Как изобразить дерево?
Топология дерева
Топология дерева — только листья, узлы, (корень)
Как изобразить дерево?
Топология дерева
Топология дерева — только листья, узлы, (корень)
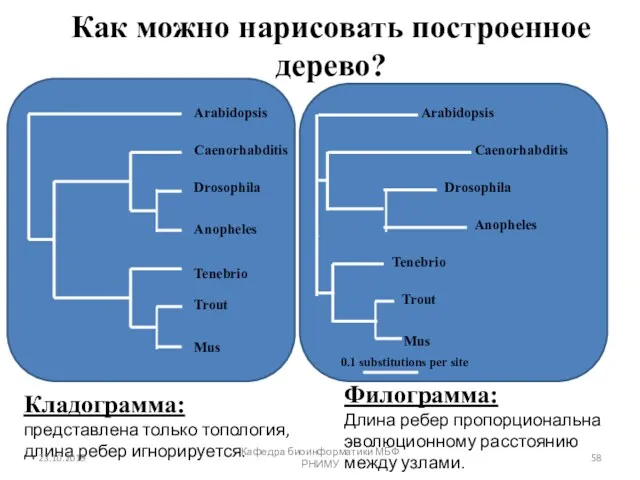
Филограмма:
Длина ребер пропорциональна эволюционному расстоянию между узлами.
Кладограмма:
представлена только топология, длина
Филограмма:
Длина ребер пропорциональна эволюционному расстоянию между узлами.
Кладограмма:
представлена только топология, длина

Достоверность топологии. Bootstraps
Создадим псевдоданные:
N множественных выравниваний той же длины,
Достоверность топологии. Bootstraps
Создадим псевдоданные:
N множественных выравниваний той же длины,

Какие on-line программы строят деревья?
ClustalW. “Tree type” – nj, phylip: строит
Какие on-line программы строят деревья?
ClustalW. “Tree type” – nj, phylip: строит

Phylip
23.10.2019
Кафедра биоинформатики МБФ РНИМУ
Phylip
23.10.2019
Кафедра биоинформатики МБФ РНИМУ

Пакет Phylip
protdist — оценка эволюционных расстояний между белковыми последовательностями (вход —
Пакет Phylip
protdist — оценка эволюционных расстояний между белковыми последовательностями (вход —

Bootstrapping with Phylip
Надо выбрать Bootstrap options в protdist, выставить не менее
Bootstrapping with Phylip
Надо выбрать Bootstrap options в protdist, выставить не менее

Общий план действий с пакетом Phylip
Множественное выравнивание -> protdist
Bootstrap options -
Общий план действий с пакетом Phylip
Множественное выравнивание -> protdist
Bootstrap options -
 Картофель. Сорт-стандарт Невский
Картофель. Сорт-стандарт Невский размножение и спорообразование бактерий
размножение и спорообразование бактерий История зоологии. Систематические категории животного мира и отличия животных от растений
История зоологии. Систематические категории животного мира и отличия животных от растений Тренинг по борьбе с курением: Скажем сигарете - НЕТ! (Презентация)
Тренинг по борьбе с курением: Скажем сигарете - НЕТ! (Презентация) Щитовидная, паращитовидная, поджелудочная железы, надпочечники
Щитовидная, паращитовидная, поджелудочная железы, надпочечники Многообразие растений. Высшие споровые растения. Часть 2
Многообразие растений. Высшие споровые растения. Часть 2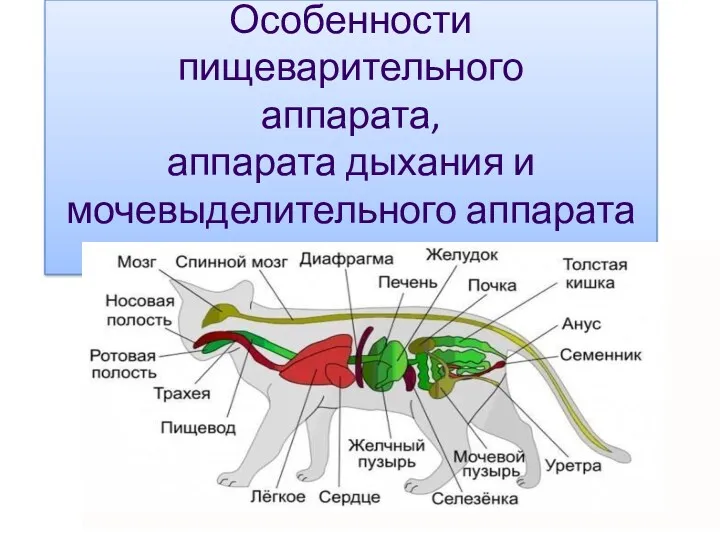 Аппарат пищеварения
Аппарат пищеварения 20231111_organ_zreniya
20231111_organ_zreniya Использование вегетативного размножения растений человеком
Использование вегетативного размножения растений человеком Семейство крестоцветные
Семейство крестоцветные Вегетативный орган высших растений побег
Вегетативный орган высших растений побег Как видят животные
Как видят животные Обитатели водоёмов
Обитатели водоёмов Эндокринная система
Эндокринная система Введение в курс физиологии. Основные принципы формирования и регуляции физиологических функций. Природа возбуждения и торможения
Введение в курс физиологии. Основные принципы формирования и регуляции физиологических функций. Природа возбуждения и торможения Тип Кольчатые черви
Тип Кольчатые черви Неживая и живая природа. (2 класс)
Неживая и живая природа. (2 класс) Растения культурные и дикорастущие
Растения культурные и дикорастущие Невидимые нити в весеннем лесу. 2 класс
Невидимые нити в весеннем лесу. 2 класс 10 интересных фактов о животных
10 интересных фактов о животных Копытные. Анатомическая особенность пальцев овец
Копытные. Анатомическая особенность пальцев овец Млекопитающие
Млекопитающие Энергетический и пластический обмен
Энергетический и пластический обмен Гормоны. Виды и классификация гормонов
Гормоны. Виды и классификация гормонов Становление систематики. 9 класс
Становление систематики. 9 класс Обитатели почвы
Обитатели почвы Семейства покрытосеменных (цветковых ) растений
Семейства покрытосеменных (цветковых ) растений Строение клетки ткани
Строение клетки ткани