- Genome annotation

Содержание
- 2. General pipeline Raw reads
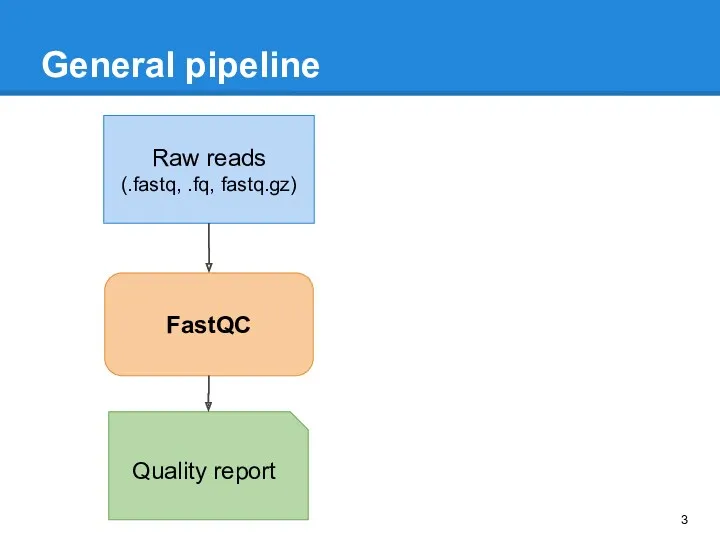
- 3. General pipeline Raw reads (.fastq, .fq, fastq.gz) FastQC Quality report
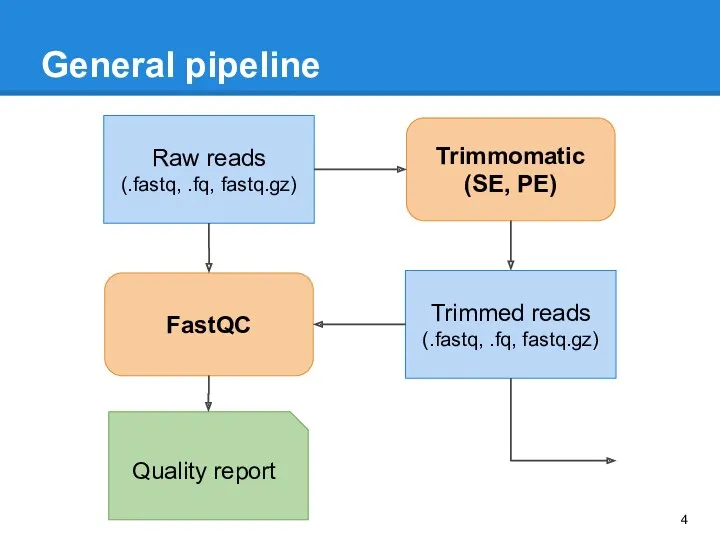
- 4. General pipeline Raw reads (.fastq, .fq, fastq.gz) FastQC Trimmomatic (SE, PE) Trimmed reads (.fastq, .fq, fastq.gz)
- 5. General pipeline Trimmed reads (.fastq, .fq, fastq.gz)
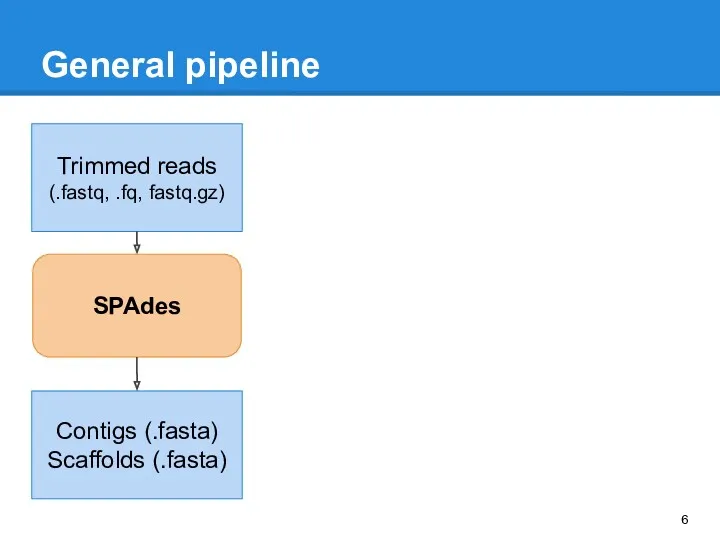
- 6. General pipeline Trimmed reads (.fastq, .fq, fastq.gz) SPAdes Contigs (.fasta) Scaffolds (.fasta)
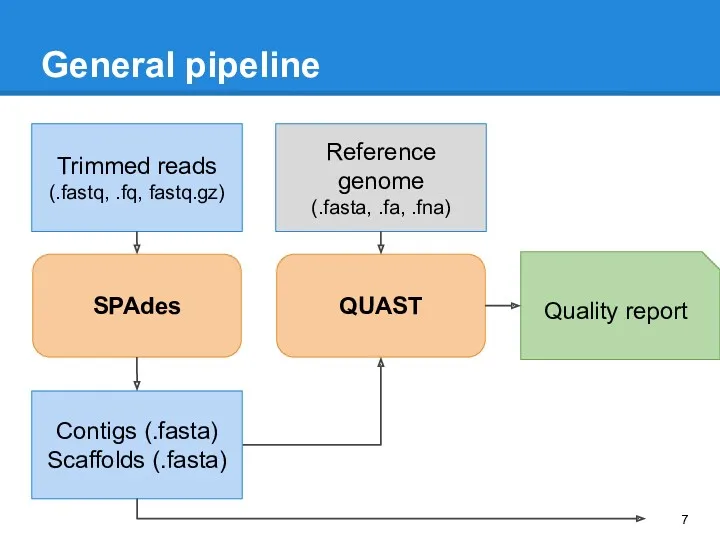
- 7. General pipeline QUAST Trimmed reads (.fastq, .fq, fastq.gz) Quality report SPAdes Contigs (.fasta) Scaffolds (.fasta) Reference
- 8. General pipeline Prokka Gene annotation (.gff, gtf) Contigs (.fasta) Scaffolds (.fasta)
- 9. Genome Annotation Questions What is the order are the genes and does this have any significance?
- 10. After completing the human genome we faced 3 Gigabytes of this: Genome sequence does not give
- 11. Not immediately apparent where the genes are…
- 12. Genomic Features Protein coding genes. In long open reading frames ORFs interrupted by introns in eukaryotes
- 13. Genome annotation STRUCTURAL ANNOTATION Open reading frame and their localization Exons, introns, UTRs Start/Stop Location of
- 14. Structural annotation Open reading frame and their localization ORFfinder, personal scripts Exons, introns, UTRs, Start/Stop, Splice
- 15. Similarity based Alignment of the known protein coding genes to contigs Will miss proteins not in
- 16. Pipeline for ideal annotation
- 17. Useful databases and web-browsers EnsEMBL -http://www.ensembl.org/index.html Vega (Vertebrate and Genome Annotation) - http://vega.sanger.ac.uk/index.html UCSC Genome Browser
- 18. Useful online annotation pipelines NCBI Prokaryotic Genomes Automatic Annotation Pipeline. - http://www.ncbi.nlm....nnotation_prok/ IGS Prokaryotic Annotation Pipeline
- 19. Bacterial genome annotation
- 20. Eukaryote vs Prokaryote Genomes
- 21. Eukaryote vs Prokaryote Genomes
- 22. Prokaryotic Genes ATG is main start codon, but GTG and TTG are also common start codons
- 23. Bacterial feature types protein coding genes promoter (-10, -35) ribosome binding site (RBS) coding sequence (CDS)
- 24. Gene-finding in Prokaryotes: Easy? ….or not? ORF Finder Open reading frame (ORF) from methionine codon to
- 25. Gene-finding in Prokaryotes: Improving predictions… Common way to search by content build Markov models of coding
- 26. Another existing tools for genome annotation:
- 27. https://www.basys.ca/
- 28. designed for Bacteria, Archaea and Viruses. It can't handle multi-exon gene models your own custom "trusted"
- 29. Prokka: rapid prokaryotic genome annotation
- 30. Prokka output .fna FASTA file of original input contigs (nucleotide) .faa FASTA file of translated coding
- 31. Prokka prokka --help prokka --docs Show full manual/documentation prokka --setupdb prokka --listdb List all configured databases
- 32. GFF - General Feature Format (V2, V2.5, V3) Designed as a single line record for describing
- 33. GFF-version 3 GROUP tag different for ALL versions GFF2: group is a unique description, usually the
- 34. GFF-version 3 GFF3: New tag “Parent” – nested multilevel structure ctg123 . gene 1000 9000 .
- 35. GFF-version 3 GFF3: FASTA seqs can be embedded
- 36. Integrative Genomics Viewer (IGV) http://software.broadinstitute.org/software/igv/home
- 37. genome viewer Artemis Free genome browser and annotation tool that allows visualization of sequence features, next
- 39. Скачать презентацию

General pipeline
Raw reads
General pipeline
Raw reads
General pipeline
Raw reads
(.fastq, .fq, fastq.gz)
FastQC
Quality report
General pipeline
Raw reads
(.fastq, .fq, fastq.gz)
FastQC
Quality report
General pipeline
Raw reads
(.fastq, .fq, fastq.gz)
FastQC
Trimmomatic
(SE, PE)
Trimmed reads
(.fastq, .fq, fastq.gz)
Quality
General pipeline
Raw reads
(.fastq, .fq, fastq.gz)
FastQC
Trimmomatic
(SE, PE)
Trimmed reads
(.fastq, .fq, fastq.gz)
Quality

General pipeline
Trimmed reads
(.fastq, .fq, fastq.gz)
General pipeline
Trimmed reads
(.fastq, .fq, fastq.gz)
General pipeline
Trimmed reads
(.fastq, .fq, fastq.gz)
SPAdes
Contigs (.fasta)
Scaffolds (.fasta)
General pipeline
Trimmed reads
(.fastq, .fq, fastq.gz)
SPAdes
Contigs (.fasta)
Scaffolds (.fasta)
General pipeline
QUAST
Trimmed reads
(.fastq, .fq, fastq.gz)
Quality report
SPAdes
Contigs (.fasta)
Scaffolds (.fasta)
Reference genome
(.fasta, .fa,
General pipeline
QUAST
Trimmed reads
(.fastq, .fq, fastq.gz)
Quality report
SPAdes
Contigs (.fasta)
Scaffolds (.fasta)
Reference genome
(.fasta, .fa,
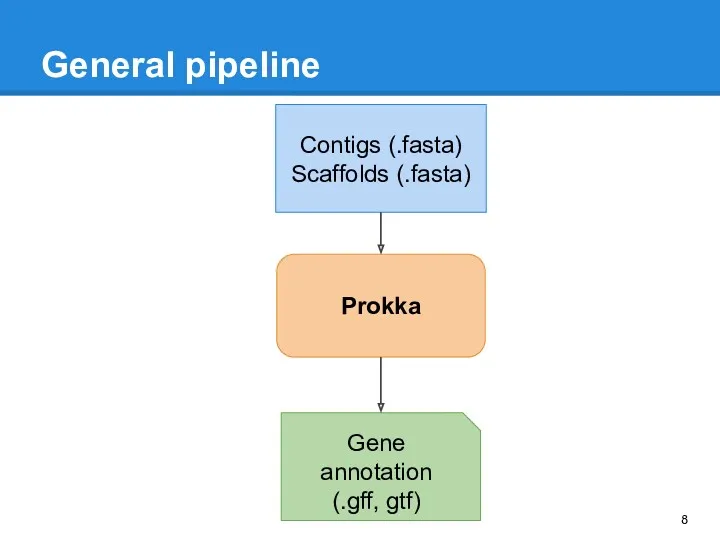
General pipeline
Prokka
Gene annotation
(.gff, gtf)
Contigs (.fasta)
Scaffolds (.fasta)
General pipeline
Prokka
Gene annotation
(.gff, gtf)
Contigs (.fasta)
Scaffolds (.fasta)

Genome Annotation Questions
What is the order are the genes and does
Genome Annotation Questions
What is the order are the genes and does

After completing the human genome we faced 3 Gigabytes of this:
Genome
After completing the human genome we faced 3 Gigabytes of this:
Genome

Not immediately apparent where the genes are…
Not immediately apparent where the genes are…

Genomic Features
Protein coding genes.
In long open reading frames
ORFs interrupted by
Genomic Features
Protein coding genes.
In long open reading frames
ORFs interrupted by
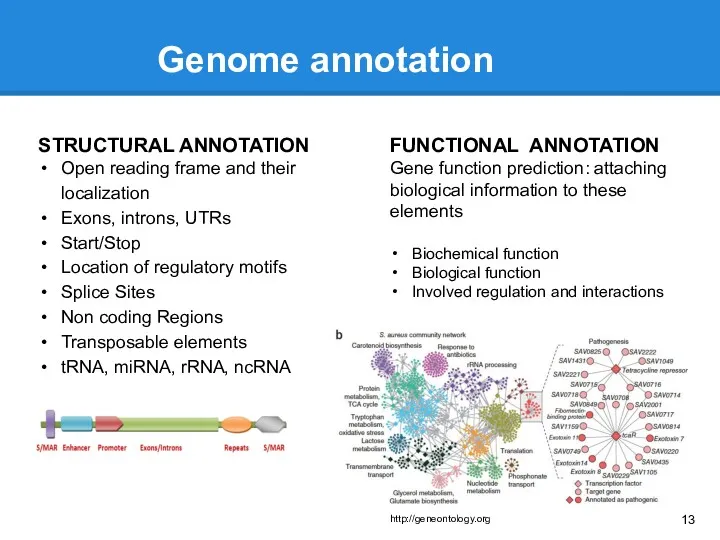
Genome annotation
STRUCTURAL ANNOTATION
Open reading frame and their localization
Exons, introns, UTRs
Start/Stop
Location of
Genome annotation
STRUCTURAL ANNOTATION
Open reading frame and their localization
Exons, introns, UTRs
Start/Stop
Location of

Structural annotation
Open reading frame and their localization
ORFfinder, personal scripts
Exons, introns,
Structural annotation
Open reading frame and their localization
ORFfinder, personal scripts
Exons, introns,

Similarity based
Alignment of the known protein coding genes to contigs
Will miss
Similarity based
Alignment of the known protein coding genes to contigs
Will miss
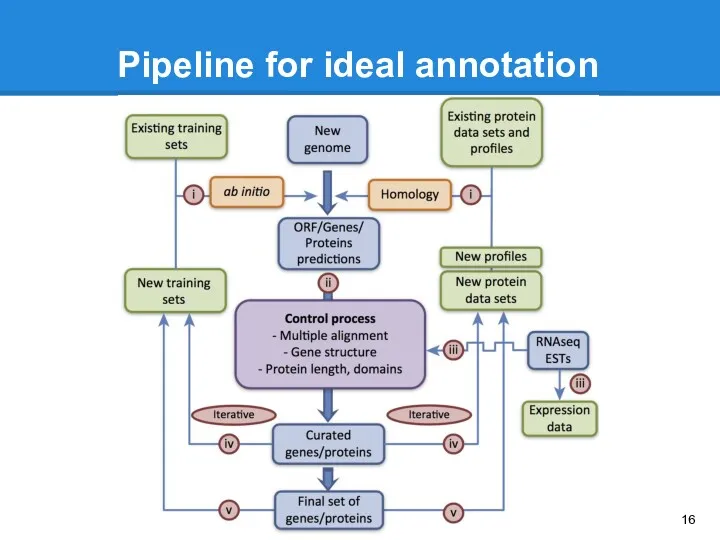
Pipeline for ideal annotation
Pipeline for ideal annotation

Useful databases and web-browsers
EnsEMBL -http://www.ensembl.org/index.html
Vega (Vertebrate and Genome Annotation) - http://vega.sanger.ac.uk/index.html
UCSC Genome Browser - http://genome.ucsc.edu/
MGC
Useful databases and web-browsers
EnsEMBL -http://www.ensembl.org/index.html
Vega (Vertebrate and Genome Annotation) - http://vega.sanger.ac.uk/index.html
UCSC Genome Browser - http://genome.ucsc.edu/
MGC

Useful online annotation pipelines
NCBI Prokaryotic Genomes Automatic Annotation Pipeline. - http://www.ncbi.nlm....nnotation_prok/
IGS Prokaryotic
Useful online annotation pipelines
NCBI Prokaryotic Genomes Automatic Annotation Pipeline. - http://www.ncbi.nlm....nnotation_prok/
IGS Prokaryotic

Bacterial genome annotation
Bacterial genome annotation
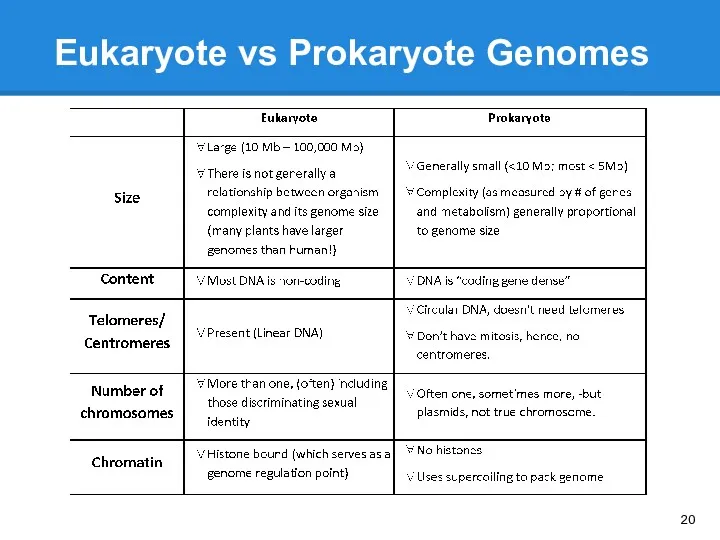
Eukaryote vs Prokaryote Genomes
Eukaryote vs Prokaryote Genomes
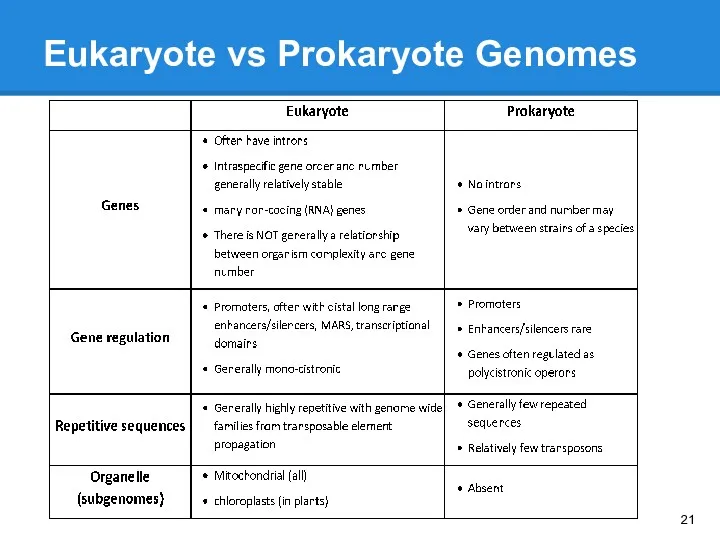
Eukaryote vs Prokaryote Genomes
Eukaryote vs Prokaryote Genomes

Prokaryotic Genes
ATG is main start codon, but GTG and TTG
Prokaryotic Genes
ATG is main start codon, but GTG and TTG

Bacterial feature types
protein coding genes
promoter (-10, -35)
ribosome binding site (RBS)
coding sequence
Bacterial feature types
protein coding genes
promoter (-10, -35)
ribosome binding site (RBS)
coding sequence

Gene-finding in Prokaryotes:
Easy? ….or not?
ORF Finder
Open reading frame (ORF) from methionine
Gene-finding in Prokaryotes:
Easy? ….or not?
ORF Finder
Open reading frame (ORF) from methionine

Gene-finding in Prokaryotes:
Improving predictions…
Common way to search by content
build Markov models
Gene-finding in Prokaryotes:
Improving predictions…
Common way to search by content
build Markov models
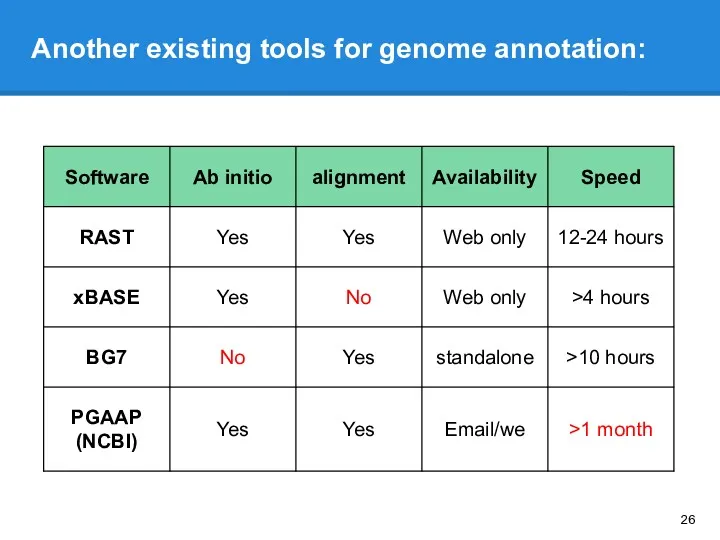
Another existing tools for genome annotation:
Another existing tools for genome annotation:
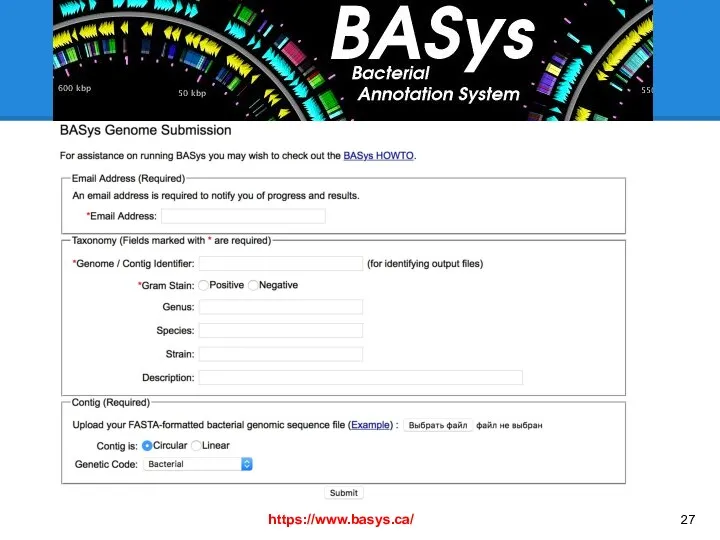
https://www.basys.ca/
https://www.basys.ca/

designed for Bacteria, Archaea and Viruses. It can't handle multi-exon gene
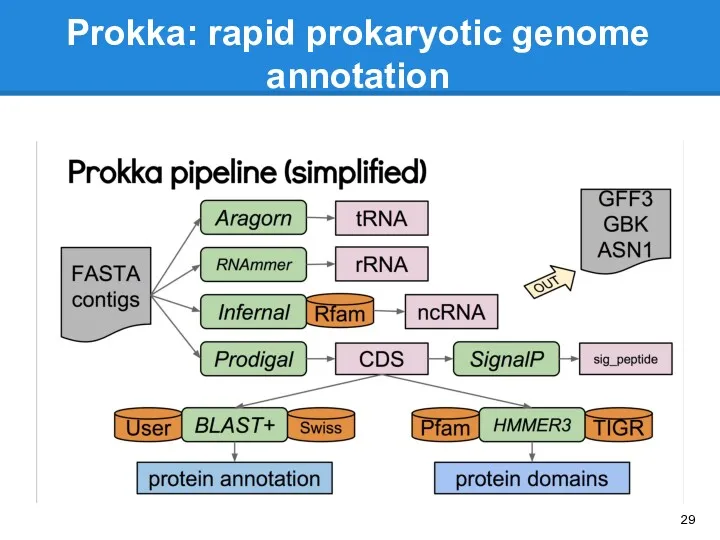
Prokka: rapid prokaryotic genome annotation
Prokka: rapid prokaryotic genome annotation

Prokka output
.fna FASTA file of original input contigs (nucleotide)
.faa FASTA
Prokka output
.fna FASTA file of original input contigs (nucleotide)
.faa FASTA

Prokka
prokka --help
prokka --docs Show full manual/documentation
prokka --setupdb
prokka --listdb List all
Prokka
prokka --help
prokka --docs Show full manual/documentation
prokka --setupdb
prokka --listdb List all

GFF - General Feature Format (V2, V2.5, V3)
Designed as a single
GFF - General Feature Format (V2, V2.5, V3)
Designed as a single

GFF-version 3
GROUP tag different for ALL versions
GFF2: group is a unique
GFF-version 3
GROUP tag different for ALL versions
GFF2: group is a unique

GFF-version 3
GFF3: New tag “Parent” – nested multilevel structure
ctg123 . gene 1000
GFF-version 3
GFF3: New tag “Parent” – nested multilevel structure
ctg123 . gene 1000

GFF-version 3
GFF3: FASTA seqs can be embedded
GFF-version 3
GFF3: FASTA seqs can be embedded
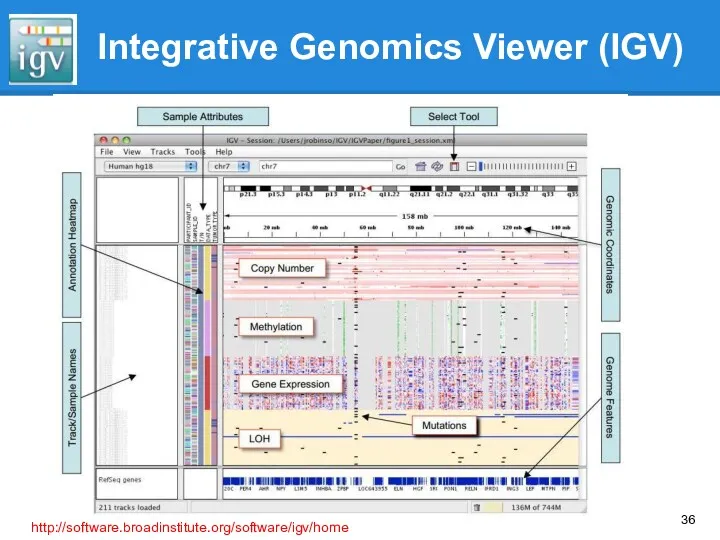
Integrative Genomics Viewer (IGV)
http://software.broadinstitute.org/software/igv/home
Integrative Genomics Viewer (IGV)
http://software.broadinstitute.org/software/igv/home
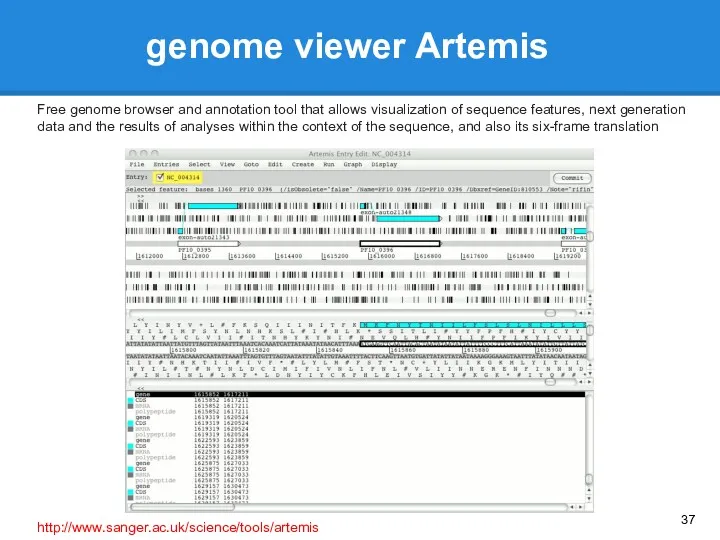
genome viewer Artemis
Free genome browser and annotation tool that allows visualization
genome viewer Artemis
Free genome browser and annotation tool that allows visualization
 Физиологический процесс дыхание
Физиологический процесс дыхание Общая характеристика надкласса Рыбы. Хрящевые рыбы
Общая характеристика надкласса Рыбы. Хрящевые рыбы Тыныс алудың организм үшін маңызы. Тыныс алу кезеңдері. Тыныс алудың реттелуі
Тыныс алудың организм үшін маңызы. Тыныс алу кезеңдері. Тыныс алудың реттелуі Ядовитые растения
Ядовитые растения Введение в клеточную биологию. Свойства живого. Клеточная теория
Введение в клеточную биологию. Свойства живого. Клеточная теория Надцарство актиномицеты
Надцарство актиномицеты Характеристика іонізуючих випромінювань і взаємодія їх із речовиною
Характеристика іонізуючих випромінювань і взаємодія їх із речовиною Зуби ссавців
Зуби ссавців Урок биологии в 9 классе по теме Антропогенез
Урок биологии в 9 классе по теме Антропогенез Селекция растений
Селекция растений Антропогенез. Расы. Расизм. Часть 5
Антропогенез. Расы. Расизм. Часть 5 Клиническая анатомия органов малого таза у женщин
Клиническая анатомия органов малого таза у женщин Исчезающие виды птиц в Самарской области
Исчезающие виды птиц в Самарской области Слюнные железы
Слюнные железы Структурно-функциональная организация клетки
Структурно-функциональная организация клетки Класс Пресмыкающиеся, или Рептилии. Отряд Чешуйчатые
Класс Пресмыкающиеся, или Рептилии. Отряд Чешуйчатые Пищеварительная система. Микропрепараты по гистологии
Пищеварительная система. Микропрепараты по гистологии Жасуша түралы түсінік
Жасуша түралы түсінік Гюннинен 211 мутагены
Гюннинен 211 мутагены Хвощи. Внешний вид хвоща
Хвощи. Внешний вид хвоща Растительный и животный мир Антарктиды
Растительный и животный мир Антарктиды Өсімдіктер
Өсімдіктер Вегетативное размножение.
Вегетативное размножение. Лекарственные растения
Лекарственные растения Науки об организме человека
Науки об организме человека Причини вимирання динозаврів
Причини вимирання динозаврів Птицы леса
Птицы леса Презентация Биография И. Н. Сеченова, 8 кл
Презентация Биография И. Н. Сеченова, 8 кл