- Индивидуальные пути обмена аминокислот. Часть 2. Лекция №14

Содержание
- 2. Фенилаланин - незаменимая аминокислота, так как в клетках животных не синтезируется ароматическое кольцо. Основная часть поступающего
- 3. ФЕНИЛАЛАНИН ТИРОЗИН ОПК Белки Ацетоацетат Фумарат Глюкоза Катехоламины Меланины Йодтиронины Дофамин Норадреналин Адреналин Нервная ткань Надпочечники
- 4. Реакция гидроксилирования фенилаланина и регенерация тетрагидробиоптерина (Н4БП) Реакцию катализирует фенилаланингидроксилаза (1), коферментом которой является Н4БП. Кофактором
- 5. Пути превращения фенилаланина и тирозина в разных тканях
- 6. Тирозингидроксилаза (Fe2+) Адреналин Норадреналин Синтез катехоламинов (надпочечники, нервная ткань) О2 Н2О ДОФА-декарбоксилаза (В6) СО2 ДОФамингид- роксилаза
- 7. Значение катехоламинов ДОФамин – нейромедиатор среднего отдела мозга. часть системы «поощрения», вызывает чувства удовлетворения, стимулирует процессы
- 8. Значение катехоламинов Норадреналин тормозный медиатор симпатической нервной системы и разных отделов головного мозга, возбуждающий медиатор в
- 9. Значение катехоламинов Адреналин гормон интенсивной физической работы, при стрессе, регулирует основной обмен, усиливает сокращение сердечной мышцы.
- 10. Болезнь Паркинсона – развивается при гипосек-реции ДОФамина в черной субстанции мозга (в среднем отделе мозга). Частота
- 11. Синтез меланинов (меланоциты) Кожа Волосы Радужная оболочка глаз О2 Н2О Тирозиназа (Cu2+)
- 12. Синтез йодтиронинов (щитовидная железа) Тиреоглобулин 2. Тирозин 2 J - е J20 (фиксация и окисление) 1.
- 13. Синтез трийодтиронина (Т3) Монойодтирозин (3-йодтирозин) Дийодтирозин (3,5-дийодтирозин) ала Тиреоглобулин 4. Трийодтиронин (Т3) 3 3 5 3
- 14. Синтез тетрайодтиронина (тироксина, Т4) Дийодтирозин (3,5-дийодтирозин) 3 5 Дийодтирозин (3,5-дийодтирозин) 3 5 Тетрайодтиронин (тироксин, Т4) 5.
- 15. Фенилаланин Тирозин n-гидроксифенилпируват Гомогентизиновая кислота Фумарилацетоацетат Фумарат Ацетоацетат Катаболизм фенилаланина и тирозина в печени О2 Н2О
- 16. СН2 – СН – СООН NН2 Фенилаланин ПФ α - КГ Глу СН2 – С –
- 17. Белки (пищи и тканей) Фен Врожденные нарушения обмена ФЕН и ТИР Фенилпируват Фениллактат Фенилацетат Тир ДОФА
- 18. Фенилкетонурия – наследственное заболевание, наследуется по аутосомно-рецессивному типу, частота 1:10 тыс. новорожденных. дефект фермента фенилаланингидроксилазы. В
- 19. Проявления ФКУ – нарушения умственного и физического развития, судорожный синдром, нарушение пигментации. Больные не доживают до
- 20. Наследуется по аутосомно-рецессивному типу, частота 1:20 тыс. новорожденных. Причина метаболического нарушения - врожденный дефект тирозиназы, катализирующей
- 21. Наследуется по аутосомно-рецессивному типу, частота встречаемости – 2-5 : 1 млн. новорожденных. Причина заболевания - дефект
- 22. Нарушения катаболизма тирозина в печени приводит к тирозинемии и тирозинурии. Различают 3 типа тирозинемии: Тирозинемия 1
- 23. Тирозинемия 2 типа (Синдром Рихнера –Ханхорта). Причиной является дефект фермента тирозинаминотрансферазы. Для заболевания характерны: поражения глаз
- 24. Тирозинемия новорожденных – 3 типа (кратковременная). Причина – дефект фермента n–гидроксифенилпируватдиоксигеназы. В крови повышается концентрация п-гидроксифе-нилацетата,
- 25. Обмен дикарбоновых аминокислот (глу, асп – заменимые, гликогенные) ГЛУ α - КГ Оксало- ацетат АСП Глю
- 26. Глутамат является возбудительным медиатором, в процессе метаболизма превращается в тормозные нейромедиаторы Янтарный полуальдегид γ-Бутиробетаин CH3 CH3
- 27. Биологическая роль γ-аминомасляной кислоты (ГАМК) ГАМК – увеличивает проницаемость постсинаптических мембран для ионов К+, что вызывает
- 28. Биологическая роль ГЛУ + NH3 + АТФ Глутаминсинтаза АТФ + Фн Глу
- 29. Использование глутамина и аспартата для синтеза пуриновых оснований Пуриновое кольцо формируется на остатке рибозо-5-фосфата при участии
- 30. АСП – для синтеза α-ала, β-ала (β-ала - КоА, дипептды мышечной ткани – карнозин, ансерин)
- 31. Валин, лейцин, изолейцин Незаменимые аминокислоты Вал Лей Илей гликогенная (пропионил-КоА сукцинил-КоА глю) кетогенная гликокетогенная (ацетил-КоА +
- 32. Обмен аминокислот с разветвленной цепью Лейцин Изолейцин Валин α-Кетоизокапроат α-Кето-β-метилвалериат α-Кетоизовалериат Ацил-КоА- производ- ные жирных кислот
- 33. β-окисление Врожденные нарушения разветвленных аминокислот «Моча с запахом кленового сиропа» α-кетоизовалери-ановая к-та ТДФ Лей Илей α-кетоизокап-
- 34. «Моча с запахом кленового сиропа» Болезнь Кленового Сиропа (Maple Syrup Urine Disease, лейциноз, разветвлённоцепочечная кетонурия, болезнь
- 36. Скачать презентацию

Фенилаланин - незаменимая аминокислота, так как в клетках животных не
Фенилаланин - незаменимая аминокислота, так как в клетках животных не
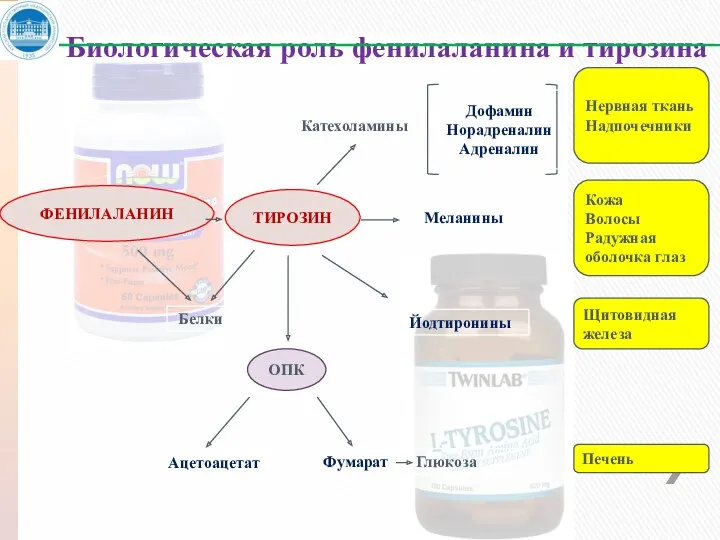
ФЕНИЛАЛАНИН
ТИРОЗИН
ОПК
Белки
Ацетоацетат
Фумарат Глюкоза
Катехоламины
Меланины
Йодтиронины
Дофамин
Норадреналин
Адреналин
Нервная ткань
Надпочечники
Кожа
Волосы
Радужная оболочка глаз
Щитовидная железа
Печень
Биологическая роль фенилаланина и
ФЕНИЛАЛАНИН
ТИРОЗИН
ОПК
Белки
Ацетоацетат
Фумарат Глюкоза
Катехоламины
Меланины
Йодтиронины
Дофамин
Норадреналин
Адреналин
Нервная ткань
Надпочечники
Кожа
Волосы
Радужная оболочка глаз
Щитовидная железа
Печень
Биологическая роль фенилаланина и
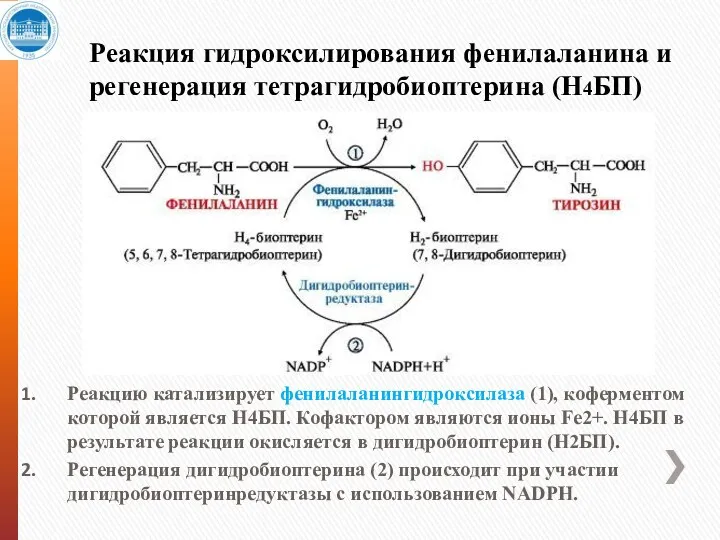
Реакция гидроксилирования фенилаланина и регенерация тетрагидробиоптерина (Н4БП)
Реакцию катализирует фенилаланингидроксилаза (1), коферментом
Реакция гидроксилирования фенилаланина и регенерация тетрагидробиоптерина (Н4БП)
Реакцию катализирует фенилаланингидроксилаза (1), коферментом
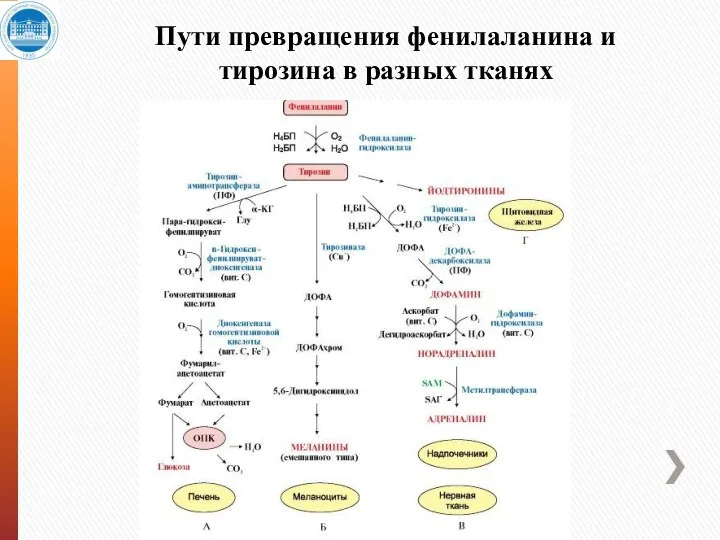
Пути превращения фенилаланина и тирозина в разных тканях
Пути превращения фенилаланина и тирозина в разных тканях
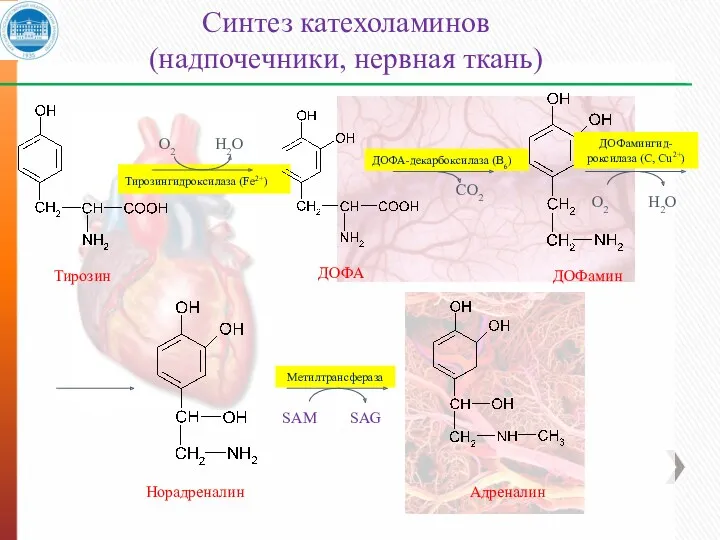
Тирозингидроксилаза (Fe2+)
Адреналин
Норадреналин
Синтез катехоламинов
(надпочечники, нервная ткань)
О2
Н2О
ДОФА-декарбоксилаза (В6)
СО2
ДОФамингид-
роксилаза (С, Сu2+)
О2
Н2О
Метилтрансфераза
SAM
SAG
Тирозингидроксилаза (Fe2+)
Адреналин
Норадреналин
Синтез катехоламинов
(надпочечники, нервная ткань)
О2
Н2О
ДОФА-декарбоксилаза (В6)
СО2
ДОФамингид-
роксилаза (С, Сu2+)
О2
Н2О
Метилтрансфераза
SAM
SAG
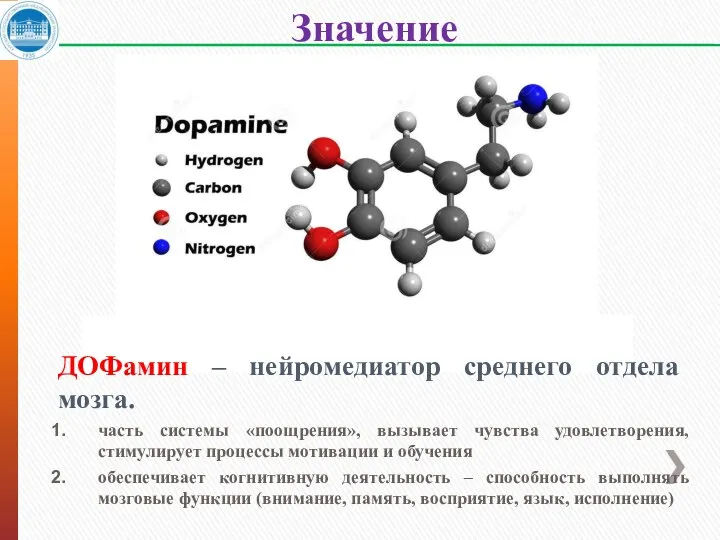
Значение катехоламинов
ДОФамин – нейромедиатор среднего отдела мозга.
часть системы «поощрения», вызывает чувства
Значение катехоламинов
ДОФамин – нейромедиатор среднего отдела мозга.
часть системы «поощрения», вызывает чувства
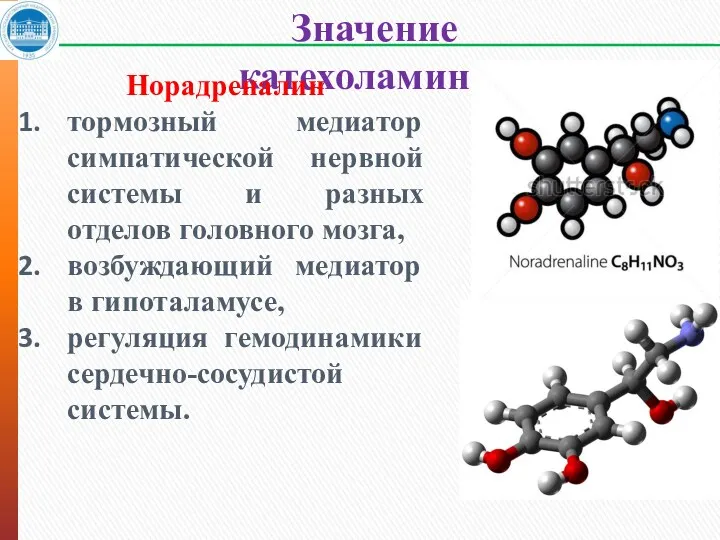
Значение катехоламинов
Норадреналин
тормозный медиатор симпатической нервной системы и разных отделов головного
Значение катехоламинов
Норадреналин
тормозный медиатор симпатической нервной системы и разных отделов головного
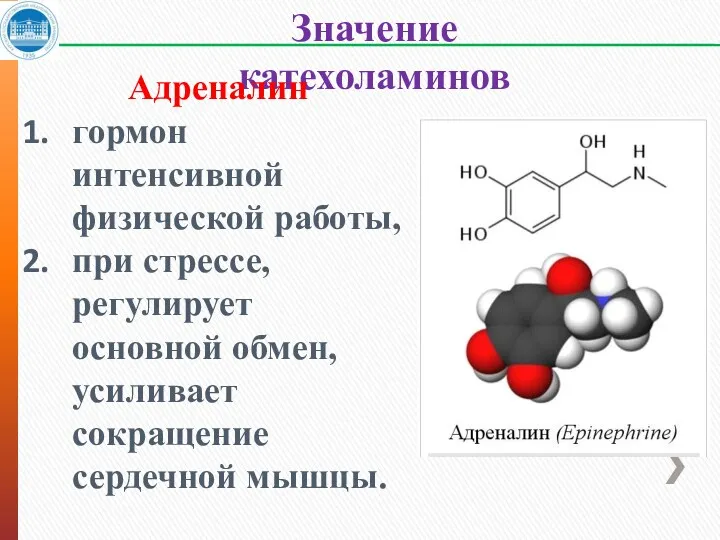
Значение катехоламинов
Адреналин
гормон интенсивной физической работы,
при стрессе, регулирует основной обмен, усиливает
Значение катехоламинов
Адреналин
гормон интенсивной физической работы,
при стрессе, регулирует основной обмен, усиливает

Болезнь Паркинсона – развивается при гипосек-реции ДОФамина в черной субстанции мозга
Болезнь Паркинсона – развивается при гипосек-реции ДОФамина в черной субстанции мозга
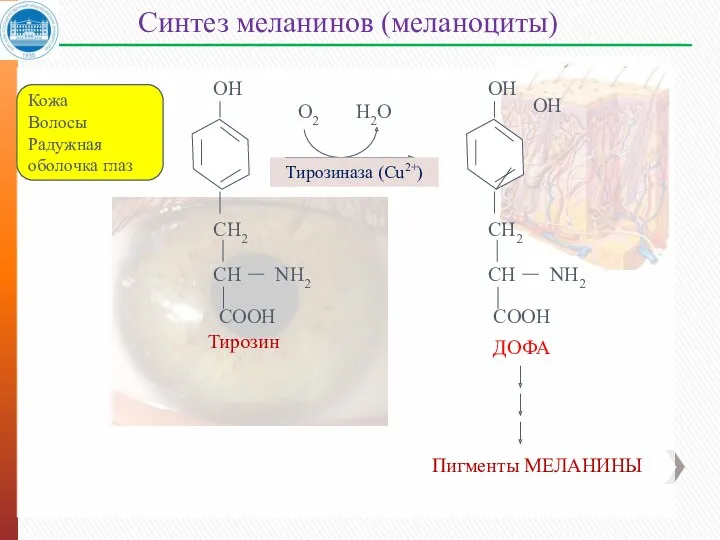
Синтез меланинов (меланоциты)
Кожа
Волосы
Радужная оболочка глаз
О2
Н2О
Тирозиназа (Cu2+)
Синтез меланинов (меланоциты)
Кожа
Волосы
Радужная оболочка глаз
О2
Н2О
Тирозиназа (Cu2+)
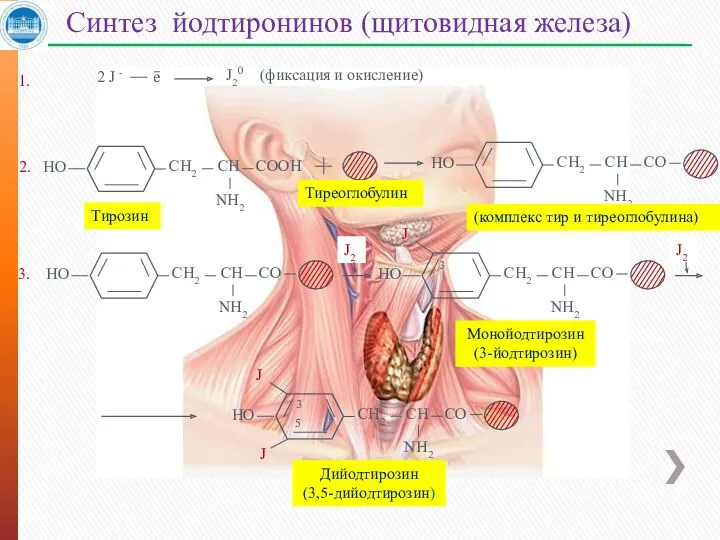
Синтез йодтиронинов (щитовидная железа)
Тиреоглобулин
2.
Тирозин
2 J -
е
J20
(фиксация и окисление)
1.
3.
(комплекс тир и тиреоглобулина)
Монойодтирозин
(3-йодтирозин)
Дийодтирозин
(3,5-дийодтирозин)
3
3
5
Синтез йодтиронинов (щитовидная железа)
Тиреоглобулин
2.
Тирозин
2 J -
е
J20
(фиксация и окисление)
1.
3.
(комплекс тир и тиреоглобулина)
Монойодтирозин
(3-йодтирозин)
Дийодтирозин
(3,5-дийодтирозин)
3
3
5
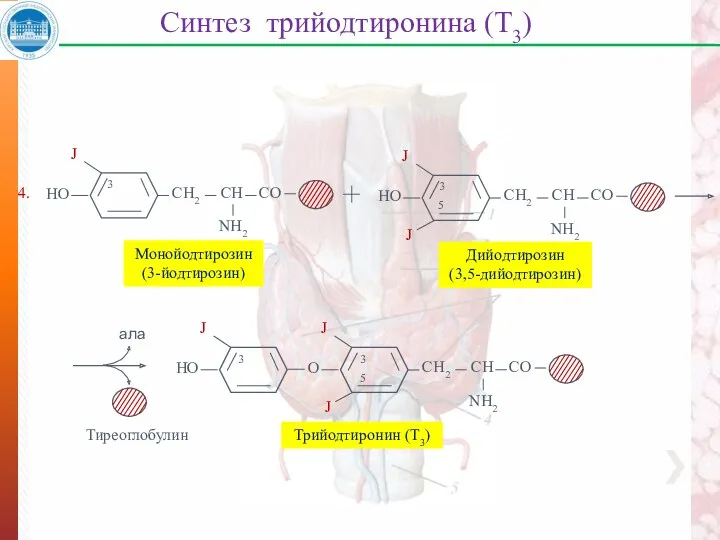
Синтез трийодтиронина (Т3)
Монойодтирозин
(3-йодтирозин)
Дийодтирозин
(3,5-дийодтирозин)
ала
Тиреоглобулин
4.
Трийодтиронин (Т3)
3
3
5
3
5
3
Синтез трийодтиронина (Т3)
Монойодтирозин
(3-йодтирозин)
Дийодтирозин
(3,5-дийодтирозин)
ала
Тиреоглобулин
4.
Трийодтиронин (Т3)
3
3
5
3
5
3
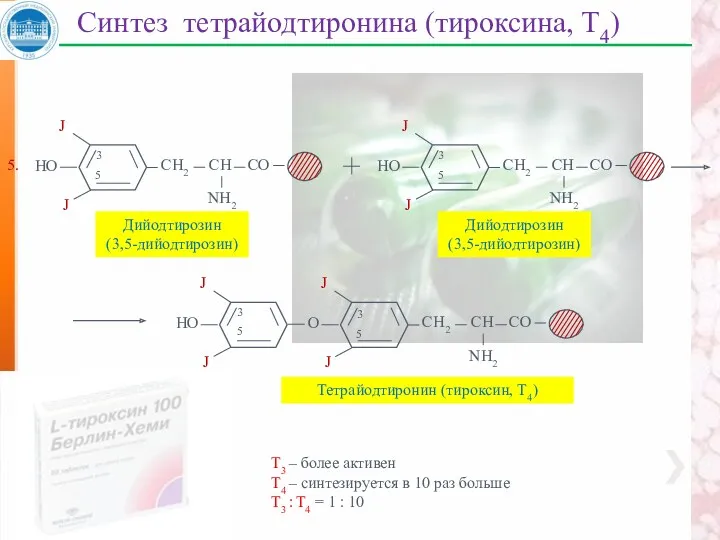
Синтез тетрайодтиронина (тироксина, Т4)
Дийодтирозин
(3,5-дийодтирозин)
3
5
Дийодтирозин
(3,5-дийодтирозин)
3
5
Тетрайодтиронин (тироксин, Т4)
5.
3
5
3
5
Т3 – более активен
Т4
Синтез тетрайодтиронина (тироксина, Т4)
Дийодтирозин
(3,5-дийодтирозин)
3
5
Дийодтирозин
(3,5-дийодтирозин)
3
5
Тетрайодтиронин (тироксин, Т4)
5.
3
5
3
5
Т3 – более активен
Т4
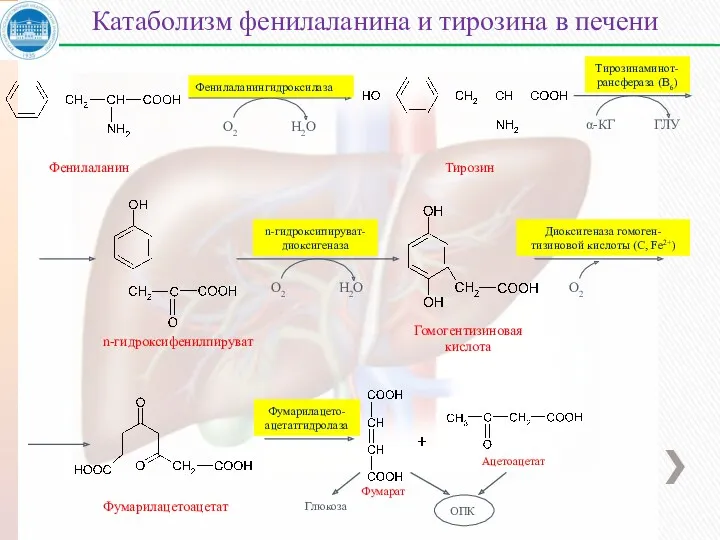
Фенилаланин
Тирозин
n-гидроксифенилпируват
Гомогентизиновая
кислота
Фумарилацетоацетат
Фумарат
Ацетоацетат
Катаболизм фенилаланина и тирозина в печени
О2
Н2О
Фенилаланингидроксилаза
α-КГ
ГЛУ
Тирозинаминот-
рансфераза (В6)
Фенилаланин
Тирозин
n-гидроксифенилпируват
Гомогентизиновая
кислота
Фумарилацетоацетат
Фумарат
Ацетоацетат
Катаболизм фенилаланина и тирозина в печени
О2
Н2О
Фенилаланингидроксилаза
α-КГ
ГЛУ
Тирозинаминот-
рансфераза (В6)
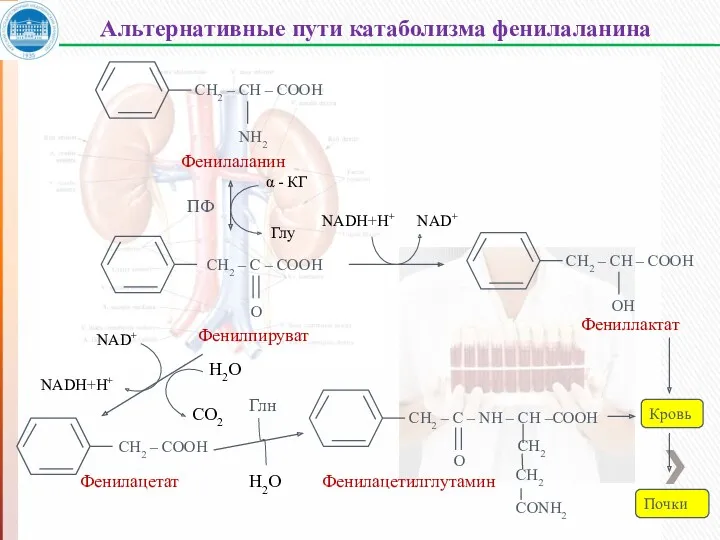
СН2 – СН – СООН
NН2
Фенилаланин
ПФ
α - КГ
Глу
СН2 – С – СООН
О
Фенилпируват
СН2
СН2 – СН – СООН
NН2
Фенилаланин
ПФ
α - КГ
Глу
СН2 – С – СООН
О
Фенилпируват
СН2
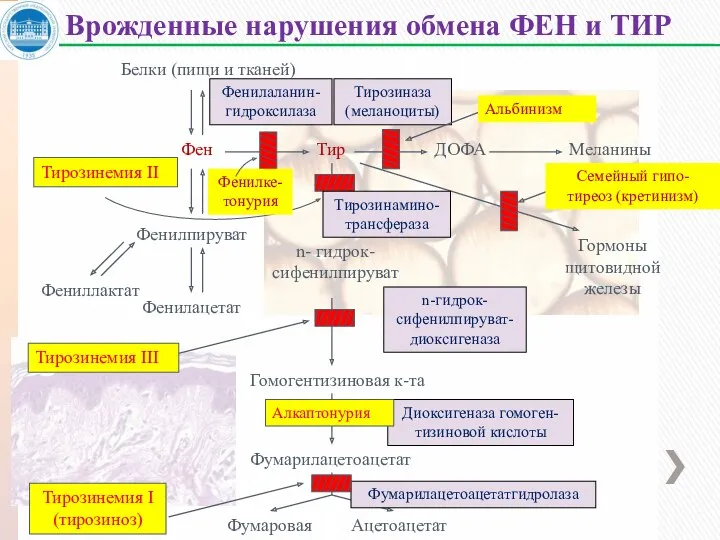
Белки (пищи и тканей)
Фен
Врожденные нарушения обмена ФЕН и ТИР
Фенилпируват
Фениллактат
Фенилацетат
Тир
ДОФА
Меланины
Гормоны
щитовидной
железы
n- гидрок-
сифенилпируват
n-гидрок-
сифенилпируват-
диоксигеназа
Фенилаланин-
гидроксилаза
Тирозиназа
(меланоциты)
Гомогентизиновая к-та
Диоксигеназа
Белки (пищи и тканей)
Фен
Врожденные нарушения обмена ФЕН и ТИР
Фенилпируват
Фениллактат
Фенилацетат
Тир
ДОФА
Меланины
Гормоны
щитовидной
железы
n- гидрок-
сифенилпируват
n-гидрок-
сифенилпируват-
диоксигеназа
Фенилаланин-
гидроксилаза
Тирозиназа
(меланоциты)
Гомогентизиновая к-та
Диоксигеназа

Фенилкетонурия – наследственное заболевание, наследуется по аутосомно-рецессивному типу, частота 1:10
Фенилкетонурия – наследственное заболевание, наследуется по аутосомно-рецессивному типу, частота 1:10

Проявления ФКУ – нарушения умственного и физического развития, судорожный синдром, нарушение
Проявления ФКУ – нарушения умственного и физического развития, судорожный синдром, нарушение

Наследуется по аутосомно-рецессивному типу, частота 1:20 тыс. новорожденных.
Причина метаболического нарушения -
Наследуется по аутосомно-рецессивному типу, частота 1:20 тыс. новорожденных.
Причина метаболического нарушения -

Наследуется по аутосомно-рецессивному типу, частота встречаемости – 2-5 : 1 млн.
Наследуется по аутосомно-рецессивному типу, частота встречаемости – 2-5 : 1 млн.
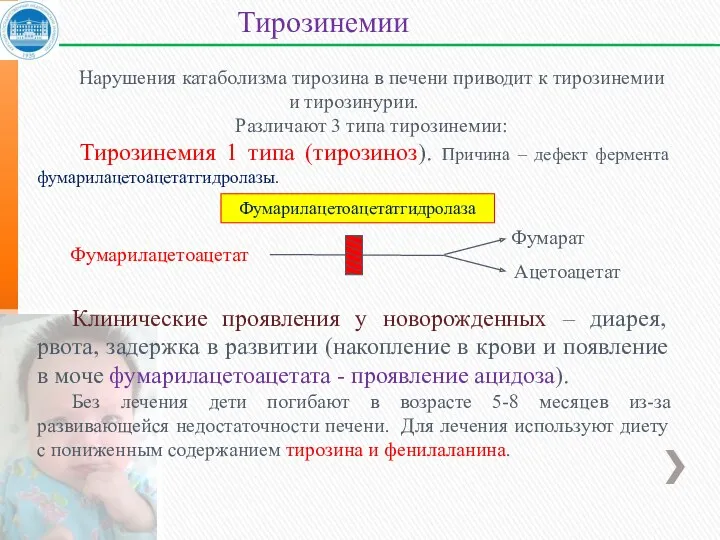
Нарушения катаболизма тирозина в печени приводит к тирозинемии и тирозинурии.
Различают
Нарушения катаболизма тирозина в печени приводит к тирозинемии и тирозинурии.
Различают

Тирозинемия 2 типа (Синдром Рихнера –Ханхорта).
Причиной является дефект фермента тирозинаминотрансферазы.
Для
Причиной является дефект фермента тирозинаминотрансферазы.
Для
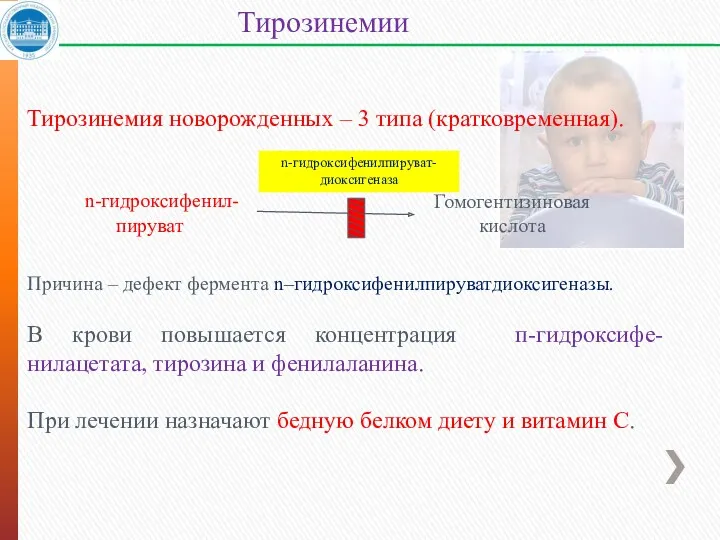
Тирозинемия новорожденных – 3 типа (кратковременная).
Причина – дефект фермента n–гидроксифенилпируватдиоксигеназы.
Тирозинемия новорожденных – 3 типа (кратковременная).
Причина – дефект фермента n–гидроксифенилпируватдиоксигеназы.
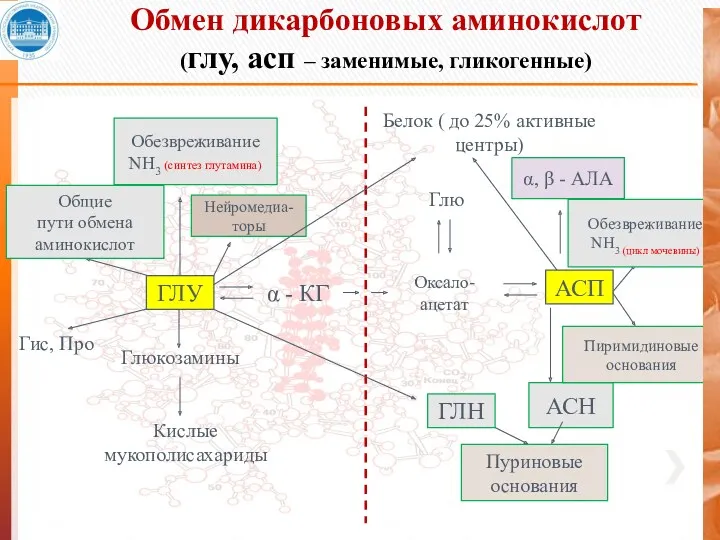
Обмен дикарбоновых аминокислот
(глу, асп – заменимые, гликогенные)
ГЛУ
α - КГ
Оксало- ацетат
АСП
Глю
Глюкозамины
Кислые
Обмен дикарбоновых аминокислот
(глу, асп – заменимые, гликогенные)
ГЛУ
α - КГ
Оксало- ацетат
АСП
Глю
Глюкозамины
Кислые
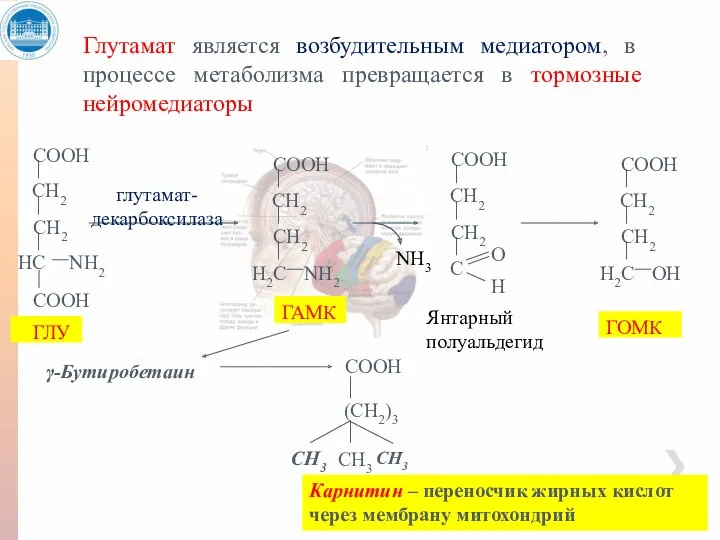
Глутамат является возбудительным медиатором, в процессе метаболизма превращается в тормозные нейромедиаторы
Глутамат является возбудительным медиатором, в процессе метаболизма превращается в тормозные нейромедиаторы

Биологическая роль
γ-аминомасляной кислоты (ГАМК)
ГАМК – увеличивает проницаемость постсинаптических мембран для ионов
Биологическая роль
γ-аминомасляной кислоты (ГАМК)
ГАМК – увеличивает проницаемость постсинаптических мембран для ионов

Биологическая роль ГЛУ
+
NH3
+
АТФ
Глутаминсинтаза
АТФ + Фн
Глу
Биологическая роль ГЛУ
+
NH3
+
АТФ
Глутаминсинтаза
АТФ + Фн
Глу
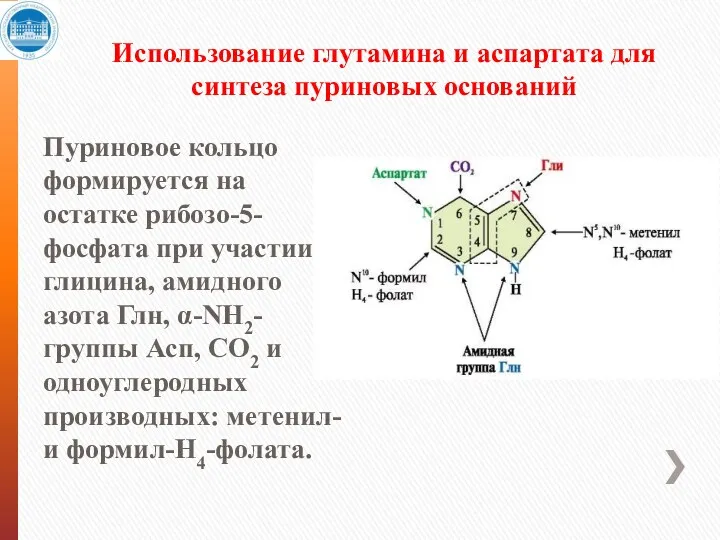
Использование глутамина и аспартата для синтеза пуриновых оснований
Пуриновое кольцо формируется на
Использование глутамина и аспартата для синтеза пуриновых оснований
Пуриновое кольцо формируется на
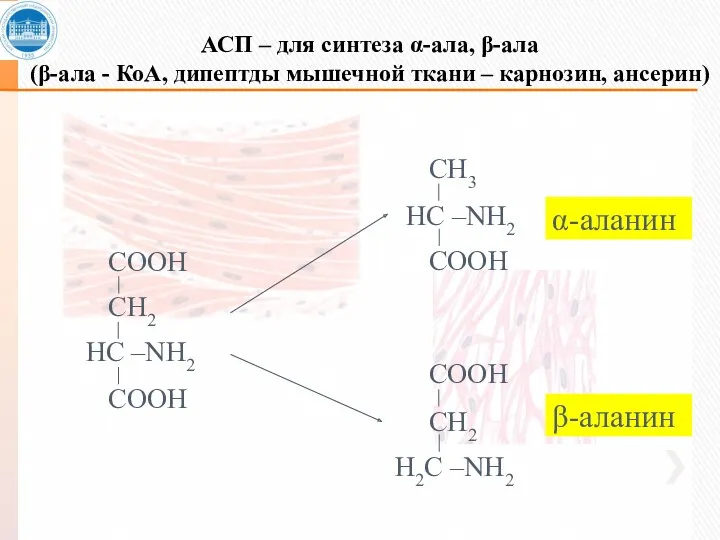
АСП – для синтеза α-ала, β-ала
(β-ала - КоА, дипептды мышечной ткани
АСП – для синтеза α-ала, β-ала (β-ала - КоА, дипептды мышечной ткани
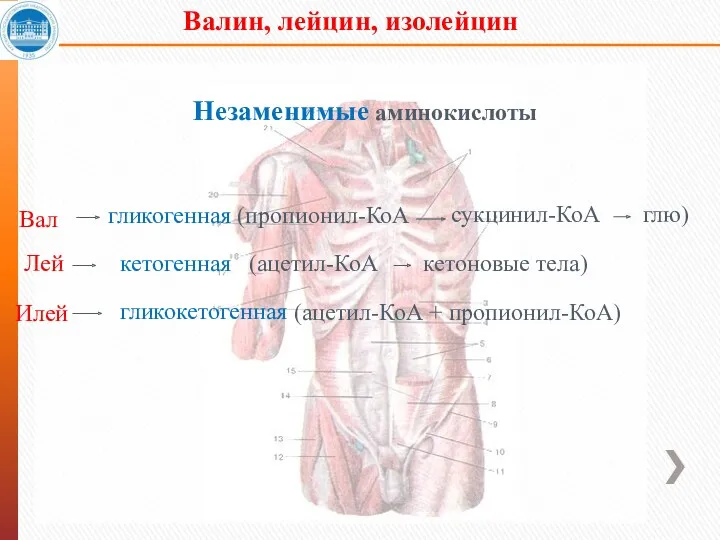
Валин, лейцин, изолейцин
Незаменимые аминокислоты
Вал
Лей
Илей
гликогенная (пропионил-КоА
сукцинил-КоА
глю)
кетогенная
гликокетогенная
(ацетил-КоА + пропионил-КоА)
(ацетил-КоА
кетоновые тела)
Валин, лейцин, изолейцин
Незаменимые аминокислоты
Вал
Лей
Илей
гликогенная (пропионил-КоА
сукцинил-КоА
глю)
кетогенная
гликокетогенная
(ацетил-КоА + пропионил-КоА)
(ацетил-КоА
кетоновые тела)
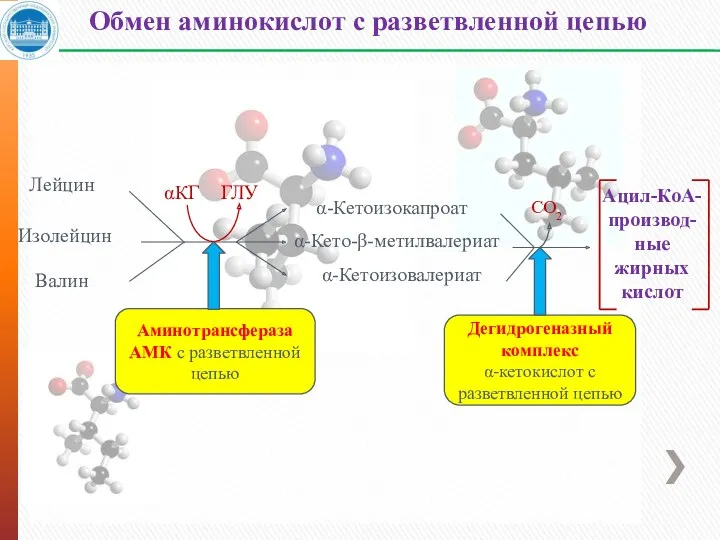
Обмен аминокислот с разветвленной цепью
Лейцин
Изолейцин
Валин
α-Кетоизокапроат
α-Кето-β-метилвалериат
α-Кетоизовалериат
Ацил-КоА-
производ-
ные
жирных
кислот
CО2
αКГ
ГЛУ
Аминотрансфераза
АМК с разветвленной цепью
Дегидрогеназный комплекс
α-кетокислот с разветвленной
Обмен аминокислот с разветвленной цепью
Лейцин
Изолейцин
Валин
α-Кетоизокапроат
α-Кето-β-метилвалериат
α-Кетоизовалериат
Ацил-КоА-
производ-
ные
жирных
кислот
CО2
αКГ
ГЛУ
Аминотрансфераза
АМК с разветвленной цепью
Дегидрогеназный комплекс
α-кетокислот с разветвленной
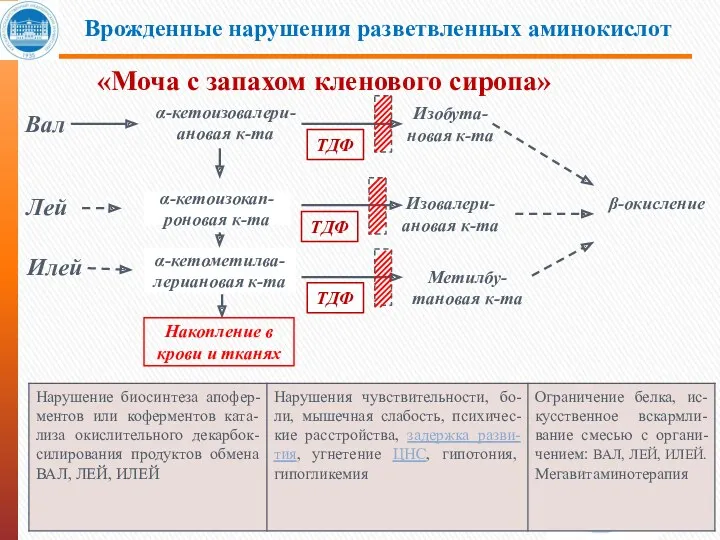
β-окисление
Врожденные нарушения разветвленных аминокислот
«Моча с запахом кленового сиропа»
α-кетоизовалери-ановая к-та
ТДФ
Лей
Илей
α-кетоизокап-
роновая к-та
α-кетометилва-
лериановая к-та
ТДФ
ТДФ
Изовалери-ановая
β-окисление
Врожденные нарушения разветвленных аминокислот
«Моча с запахом кленового сиропа»
α-кетоизовалери-ановая к-та
ТДФ
Лей
Илей
α-кетоизокап-
роновая к-та
α-кетометилва-
лериановая к-та
ТДФ
ТДФ
Изовалери-ановая

«Моча с запахом кленового сиропа»
Болезнь Кленового Сиропа (Maple Syrup Urine Disease,
«Моча с запахом кленового сиропа»
Болезнь Кленового Сиропа (Maple Syrup Urine Disease,
 Насекомые. Подготовительная группа
Насекомые. Подготовительная группа Введение в зоологию
Введение в зоологию Декоративные устройства для оформления объектов. Устройство и содержание цветников, вертикальное озеленение, каменистые участки
Декоративные устройства для оформления объектов. Устройство и содержание цветников, вертикальное озеленение, каменистые участки Тип: хордовые. Подтипы: бесчерепные и черепные, позвоночные
Тип: хордовые. Подтипы: бесчерепные и черепные, позвоночные The Human-Animal Bond
The Human-Animal Bond Топ 10 новых пород собак
Топ 10 новых пород собак В гостях у природы
В гостях у природы Парадигмы классической генетики
Парадигмы классической генетики Физиология размножения животных
Физиология размножения животных Биосинтез белка
Биосинтез белка Функции белков
Функции белков Наземно-воздушная среда обитания
Наземно-воздушная среда обитания дикий интерактив конец
дикий интерактив конец Лекарственные растения
Лекарственные растения Комнатные растения
Комнатные растения Строение и работа сердца. Круги кровообращения. Движение крови и лимфы
Строение и работа сердца. Круги кровообращения. Движение крови и лимфы Водоросли. Особенности строения, питания, размножения
Водоросли. Особенности строения, питания, размножения Регуляція експресії генів. (Лекція 2)
Регуляція експресії генів. (Лекція 2) Как появился человек на Земле
Как появился человек на Земле Презентация, конспект урока, карточки - задания к уроку биологии для 6 класса на тему Фотосинтез.
Презентация, конспект урока, карточки - задания к уроку биологии для 6 класса на тему Фотосинтез. Растительный и животный мир различных природных зон
Растительный и животный мир различных природных зон Плесневые грибы и дрожжи
Плесневые грибы и дрожжи Презентация для урока биологии. Мендель. Жизненный путь
Презентация для урока биологии. Мендель. Жизненный путь Еңбектенудің физиологиялық негізі
Еңбектенудің физиологиялық негізі Азотсодержащие гетероциклические соединения. Нуклеиновые кислоты
Азотсодержащие гетероциклические соединения. Нуклеиновые кислоты Органы чувств. Анализаторы
Органы чувств. Анализаторы Хромосомы. Набор хромосом
Хромосомы. Набор хромосом Лишайники 6 класс
Лишайники 6 класс