- Генные и хромосомные болезни

Содержание
- 2. ПЛАН ЛЕКЦИИ ВРОЖДЕННЫЕ И НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ГЕННЫЕ БОЛЕЗНИ ХРОМОСОМНЫЕ БОЛЕЗНИ МУЛЬТИФАКТОРИАЛЬНЫЕ БОЛЕЗНИ
- 3. Врожденные болезни – это группа заболеваний, с которыми новорожденный появляется на свет. Врожденные болезни могут быть
- 4. Наследственные болезни - это большая группа разнообразных заболеваний, причиной которых являются мутации – различные нарушения наследственного
- 5. Классификация наследственных болезней Классифицируются на основе генетических причин возникновения: а) генные болезни; б) хромосомные болезни; в)
- 6. Наследуемость наследственных болезней Генные болезни передаются из поколения в поколение по законам Менделя. Большинство хромосомных болезней
- 7. Летальность Много патологических аллелей приводят к летальности. Больше всего смертность проявляется на уровне зигот. Считается, что
- 9. Генетический груз Среди 1000 рожденных до 5 детей умирают в возрасте до 1 года. Все перечисленные
- 10. Распространенность наследственной патологии у детей
- 11. Наследование Аутосомно-доминантное Аутосомно-рецессивное Х-сцепленное доминантное Х-сцепленное рецессивное У-сцепленное
- 12. ГЕННЫЕ БОЛЕЗНИ
- 13. Полидактилия Клинические признаки: существует два варианта: тип А, при котором дополнительный палец функционален, и тип В,
- 16. polydactyly and sindactyly
- 17. Abnormality of structures of human organism: brachydactyly
- 18. Больной с хондродистро-фией, живший 4500 лет назад, в скульптурном изображении
- 19. Ахондроплазия - (хондродистрофия) обусловлена мутацией гена рецептора фактора роста фибробластов, вызывающей отклонения в активности некоторых ферментов
- 20. Женщина с ахондроплазией
- 21. Abnormality of structures of human organism: achondroplasia
- 22. Описано более 10 вариантов мутаций в одном гене, что обусловливает крайнюю вариабельность синдрома. Семейные случаи составляют
- 23. СИНДРОМ МАРФАНА
- 24. Серповидно-клеточная анемия: AA -(HbA HbA); Aa - (HbA HbS); aa -(HbS HbS). Эта замена обусловливает пониженную
- 26. Талассемия обусловлена мутациями глобиновых генов, приводящими к уменьшенному содержанию глобинов или полному их отсутствию. Причина а-талассемии
- 28. летальный эффект легкая форма
- 30. Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация) Аутосомно-доминантный тип наследования
- 32. Болезнь липидного обмена Болезнь Тея-Сакса. Ганглиозидоз. Наследуются по аутосомно-рецессивному типу. Ганглиозиды — это сложные гликолипиды, которые
- 34. Болезнь углеводного обмена Галактоземия. Заболевание наследуется по аутосомно-рецессивному типу. Галактоза входит в состав лактозы (молочного сахара),
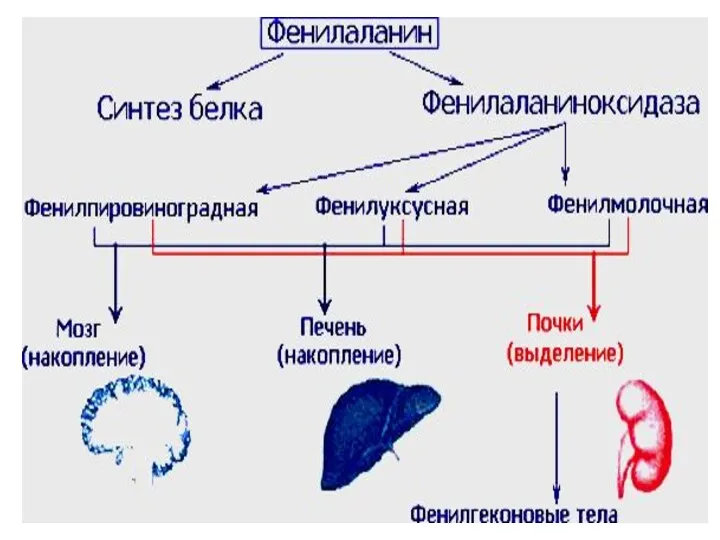
- 36. Болезни аминокислотного обмена Фенилкетонурия (фенилпировиноградная олигофрения). Заболевание, обусловлено дефектом фермента фенилаланингидроксилазы. Нарушается превращение фенилаланина в тирозин
- 37. Фенилкетонурия Клинические признаки: повышенная возбудимость и тонус мышц, тремор, «мышиный» запах, умственная отсталость, снижение образования меланина.
- 40. Альбинизм При этом заболевании в коже не образуется меланин. Первичным биохимическим дефектом является недостаточность фермента тирозиназы,
- 43. Самая большая в мире семья альбиносов насчитывает 10 человек. В довольно необычной индийской семье Пуллан насчитывается
- 45. Гемофилия А - тяжелое заболевание, обусловленное дефектом фактора VIII свертывания крови. Встречается с частотой 1:2500 новорожденных
- 46. Х – сцепленный рецессивный тип наследования (Х-Р) Гемизиготность у мужчин Заболевают преимущественно мужчины Сын никогда не
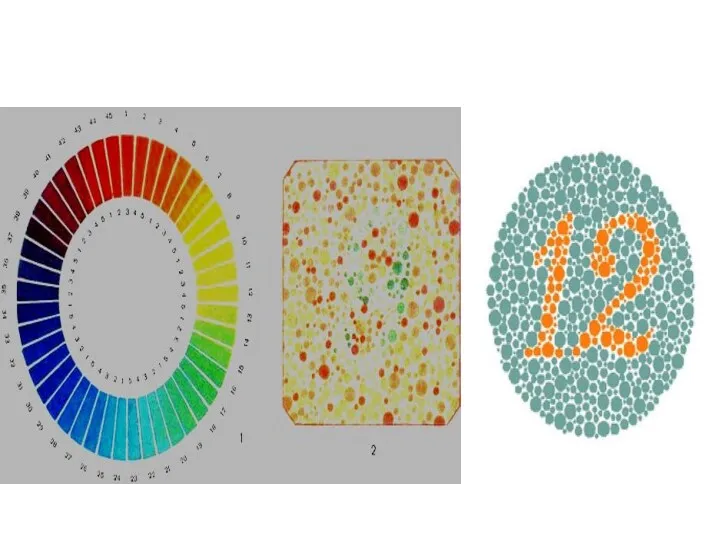
- 48. Дальтонизм
- 50. Гипофосфатемия (витамин D-резистентная форма рахита)
- 51. Х – сцепленный доминантный тип наследования (Х-Д) У здоровых родителей все дети здоровые Если отец болен
- 52. Y – сцепленный тип наследования Голандрический тип наследования Передача только по мужской линии
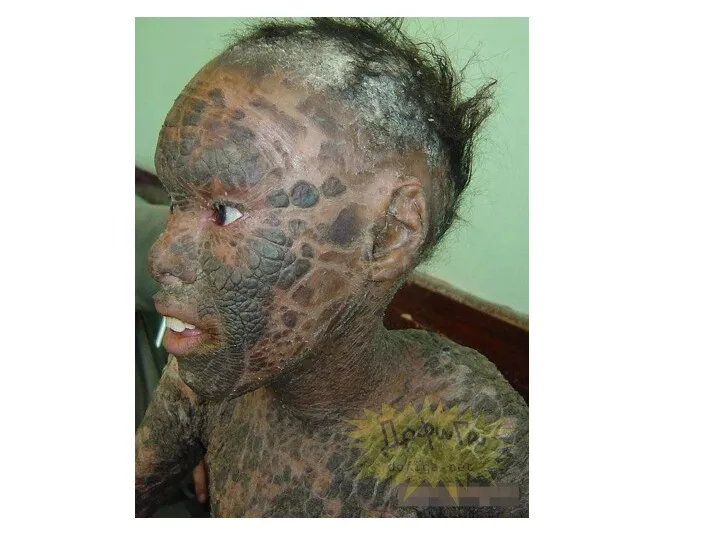
- 53. Гипертрихоз Ихтиоз- заболевание кожи. Рыбья чешуя
- 57. МУЛЬТИФАКТОРИАЛЬНЫЕ БОЛЕЗНИ С наследственной предрасположен-ностью развиваются у людей с определенным генотипом под действием факторов окружающей среды.
- 58. Примеры болезней с наследственной предрасположенностью
- 59. ШИЗОФРЕНИЯ
- 60. ХРОMOСОМНЫE БОЛЕЗНИ
- 62. СИНДРОМ КОШАЧЬЕГО КРИКА КАРИОТИП: 46, XX,( 5p-) ИЛИ 46, XY, (5p-). ЭТИОЛОГИЯ СИНДРОМА: ДЕЛЕЦИЯ КОРОТКОГО ПЛЕЧА
- 66. ФРАГИЛЬНАЯ Х-ХРОМОСОМА
- 67. ХУ СИНДРОМ МОРРИСА
- 68. СИНДРОМ ДАУНА karyotype : 47, XX, (21+) 47, XY, (21+). The frequency of births is 1/700.
- 69. Синдром Дауна Синдром - это комплекс симптомов, характерных для определенного заболевания. Синдром Дауна описан в 1866
- 70. Дети разного возраста с характерными чертами синдрома Дауна (брахицефалия, круглое лицо, макроглоссия и открытый рот, эпикант,
- 72. ТРИСОМИЯ 13 (СИНДРОМ ПАТАУ) karyotype : 47, XX, (13+) 47, XY, (13+). The frequency of births
- 73. Новорожденные с синдромом Патау. Двусторонняя расщелина верхней губы и нёба; узкие глазные щели; низко расположенные и
- 74. TRISOMY 18 (EDWARDS SYNDROME) Karyotype: 47, XX, 18+ 47, XY, 18+. Частота среди новорожденных 1/5000.
- 76. Синдром Эдвардса (трисомия 18). Смерть до 2-3 месяцев. Фенотип: узкий лоб, широкий затылок, низко расположены уши,
- 77. СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА
- 79. Девочка с синдромом Шерешевского-Тёрнера Шейные крыловидные складки; широко расположенные и недоразвитые соски молочных желёз.
- 80. Синдром Шерешевского-Тернера Причиной синдрома является отсутствие одной из Х-хромосом в женском организме. Кариотип - 45, ХО.
- 81. TРИПЛО-X СИНДРОМ Karyotype: 47, XXX. Частота среди новорожденных 1/700.
- 84. СИНДРОМ КЛАЙНФЕЛЬТЕРА Karyotype: 47, XXY. Частота среди новорожденных 1/2000 (1/500-1/700)
- 85. Синдром Клайнфельтера Синдром Клайнфельтера был описан в 1942 г. Причиной синдрома является наличие лишней Х-хромосомы в
- 86. Синдром Клайнфельтера Высокий рост, гинекомастия, женский тип оволосения на лобке.
- 87. СИНДРОМ XYY (полисомияY) karyotype : 47, XYY. Частота среди новорожденных 1/2.000.
- 88. Полисомия Y у мужчин (синдром «супермужчины»)
- 90. Механизмы появления измененных хромосомных наборов
- 91. Алкогольный синдром плода. Дети с алкогольным синдромом плода рождаются 30-45% случаев употребления алкоголя матерями. Характерные черты
- 92. Генетические болезни соматических клеток Генетические болезни соматических клеток выделены в отдельную группу в связи с обнаружением
- 93. Рак - как наследственное заболевание Среди многочисленных мульти-факториальных болезней большую группу по числу форм и частоте
- 94. Митохондриальные болезни Митохондриальные болезни — разнообразная группа заболеваний, обусловленных дефектами митохондриального генома. Выявление самостоятельных синдромов митохондриальных
- 95. Экологическая генетика Экологическая генетика человека изучает влияние факторов среды обитания на наследственность и изменчивость. Факторы окружающей
- 97. Скачать презентацию

ПЛАН ЛЕКЦИИ
ВРОЖДЕННЫЕ И НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ
ГЕННЫЕ БОЛЕЗНИ
ХРОМОСОМНЫЕ БОЛЕЗНИ
МУЛЬТИФАКТОРИАЛЬНЫЕ БОЛЕЗНИ
ПЛАН ЛЕКЦИИ
ВРОЖДЕННЫЕ И НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ
ГЕННЫЕ БОЛЕЗНИ
ХРОМОСОМНЫЕ БОЛЕЗНИ
МУЛЬТИФАКТОРИАЛЬНЫЕ БОЛЕЗНИ

Врожденные болезни – это группа заболеваний, с которыми новорожденный появляется
Врожденные болезни – это группа заболеваний, с которыми новорожденный появляется

Наследственные болезни - это большая группа разнообразных заболеваний, причиной которых являются
Наследственные болезни - это большая группа разнообразных заболеваний, причиной которых являются

Классификация наследственных болезней
Классифицируются на основе генетических причин возникновения:
а) генные
Классификация наследственных болезней
Классифицируются на основе генетических причин возникновения:
а) генные

Наследуемость наследственных болезней
Генные болезни передаются из поколения в поколение по законам
Наследуемость наследственных болезней
Генные болезни передаются из поколения в поколение по законам

Летальность
Много патологических аллелей приводят к летальности. Больше всего смертность проявляется на
Летальность
Много патологических аллелей приводят к летальности. Больше всего смертность проявляется на
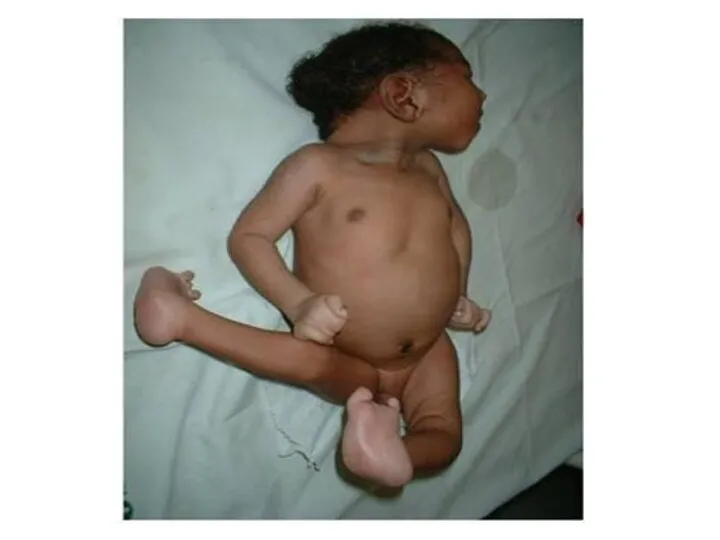

Генетический груз
Среди 1000 рожденных до 5 детей умирают в возрасте
Генетический груз
Среди 1000 рожденных до 5 детей умирают в возрасте

Распространенность наследственной патологии у детей
Распространенность наследственной патологии у детей

Наследование
Аутосомно-доминантное
Аутосомно-рецессивное
Х-сцепленное доминантное
Х-сцепленное рецессивное
У-сцепленное
Наследование
Аутосомно-доминантное
Аутосомно-рецессивное
Х-сцепленное доминантное
Х-сцепленное рецессивное
У-сцепленное
ГЕННЫЕ БОЛЕЗНИ
ГЕННЫЕ БОЛЕЗНИ
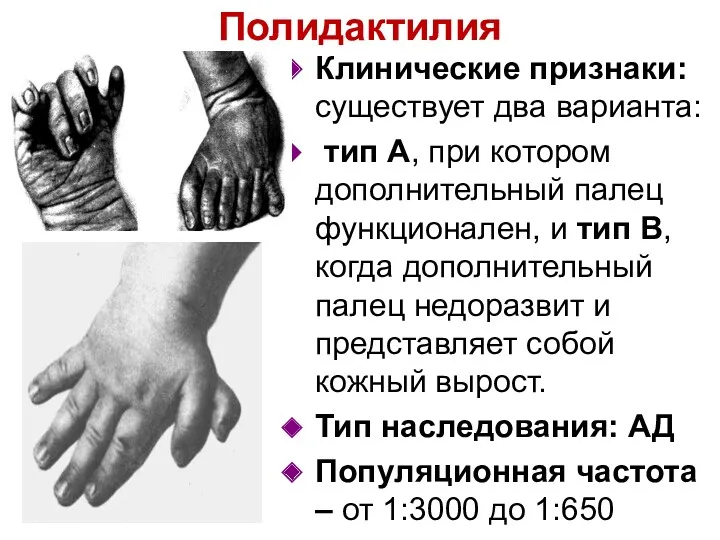
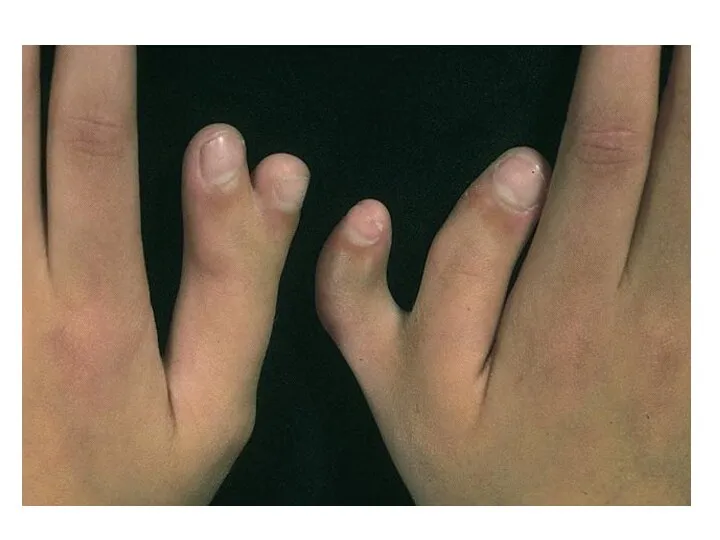
Полидактилия
Клинические признаки: существует два варианта:
тип А, при котором дополнительный палец
Полидактилия
Клинические признаки: существует два варианта:
тип А, при котором дополнительный палец
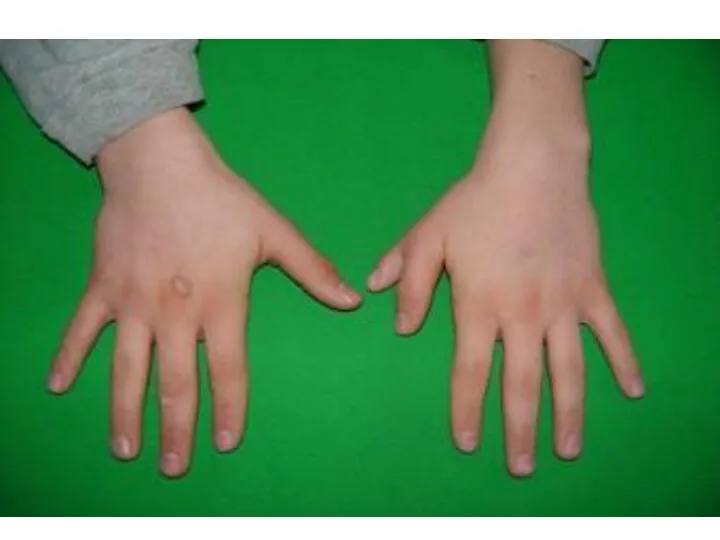
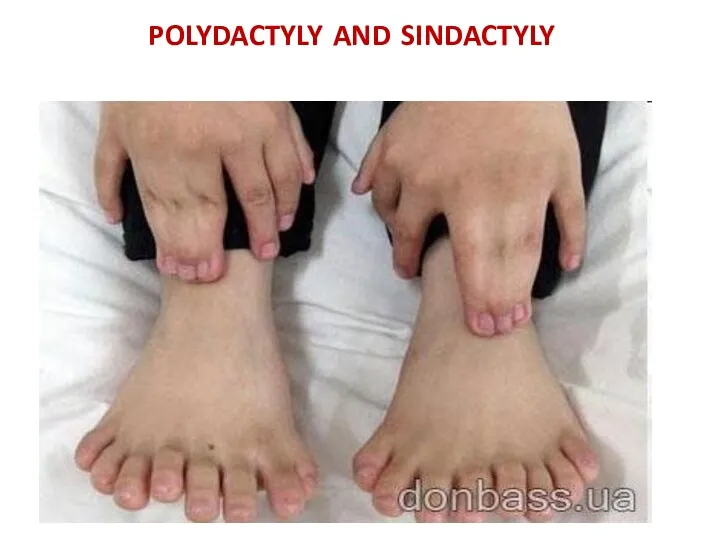
polydactyly and sindactyly
polydactyly and sindactyly
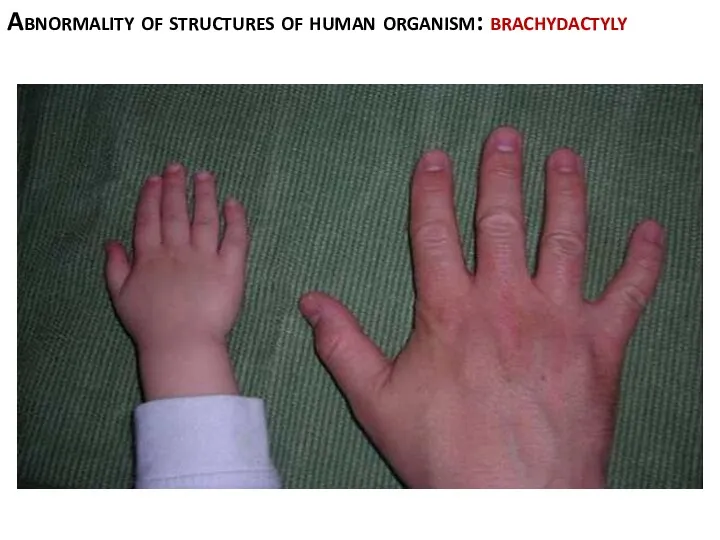
Abnormality of structures of human organism: brachydactyly
Abnormality of structures of human organism: brachydactyly

Больной с хондродистро-фией, живший 4500 лет назад, в скульптурном изображении
Больной с хондродистро-фией, живший 4500 лет назад, в скульптурном изображении

Ахондроплазия - (хондродистрофия) обусловлена мутацией гена рецептора фактора роста фибробластов,
Ахондроплазия - (хондродистрофия) обусловлена мутацией гена рецептора фактора роста фибробластов,

Женщина с ахондроплазией
Женщина с ахондроплазией

Abnormality of structures of human organism: achondroplasia
Abnormality of structures of human organism: achondroplasia

Описано более 10 вариантов мутаций в одном гене, что обусловливает
Описано более 10 вариантов мутаций в одном гене, что обусловливает
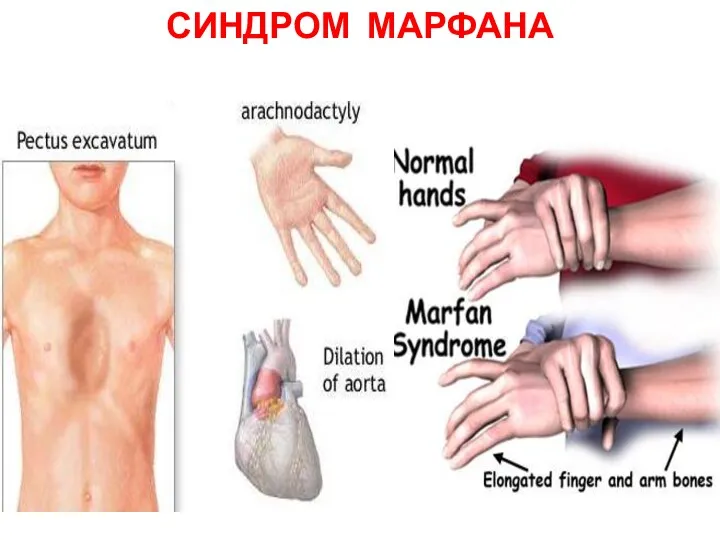
СИНДРОМ МАРФАНА
СИНДРОМ МАРФАНА
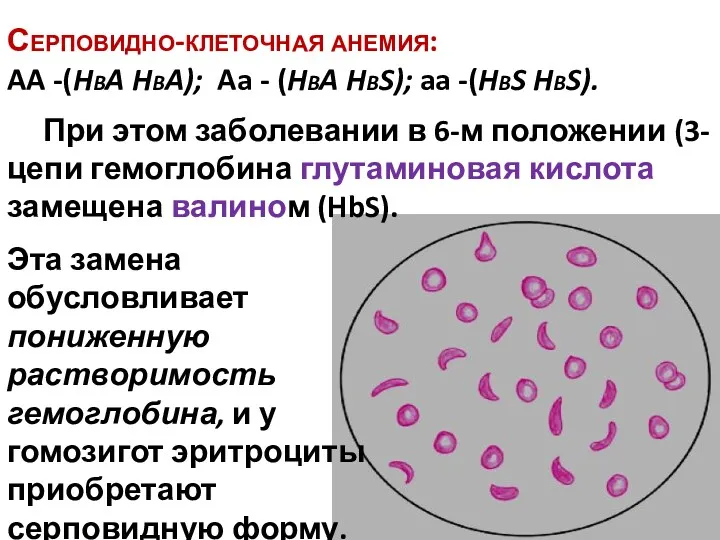
Серповидно-клеточная анемия:
AA -(HbA HbA); Aa - (HbA HbS); aa -(HbS
Серповидно-клеточная анемия: AA -(HbA HbA); Aa - (HbA HbS); aa -(HbS
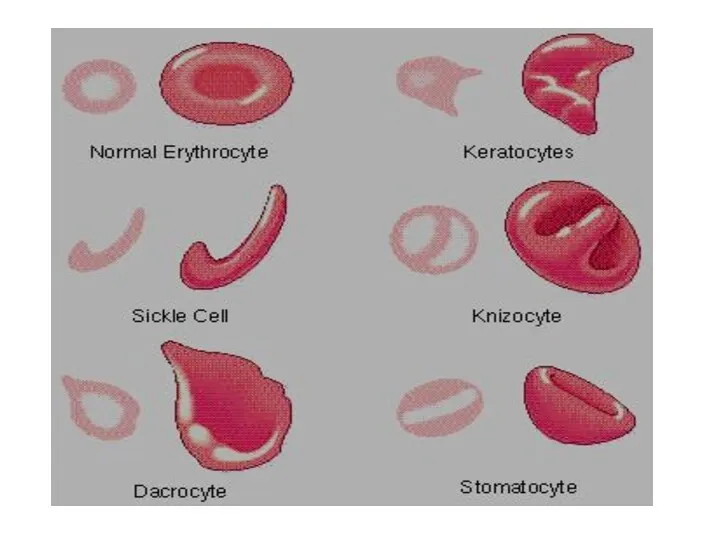

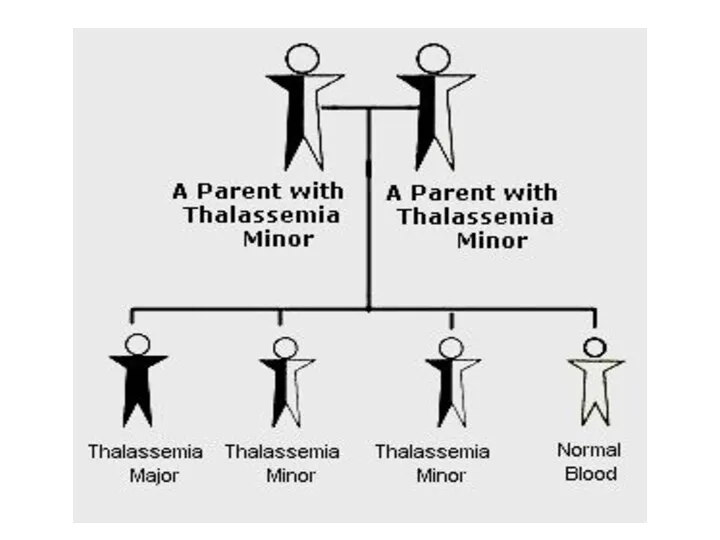
Талассемия обусловлена мутациями глобиновых генов, приводящими к уменьшенному содержанию глобинов
Талассемия обусловлена мутациями глобиновых генов, приводящими к уменьшенному содержанию глобинов
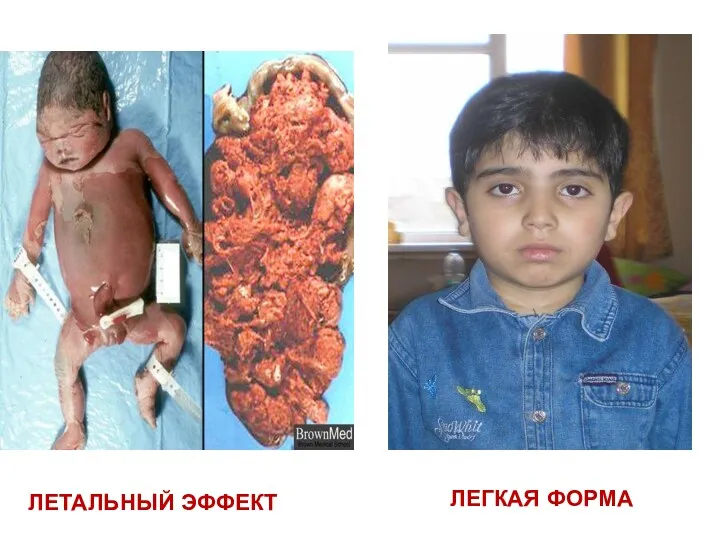
летальный эффект
легкая форма
летальный эффект
легкая форма

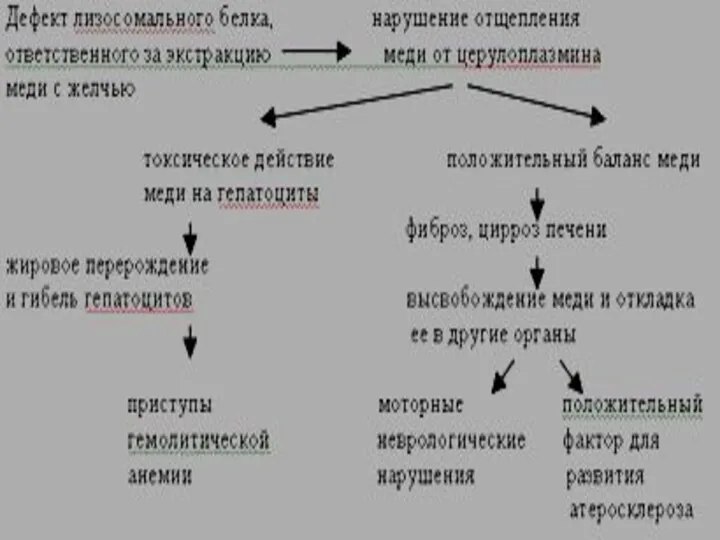
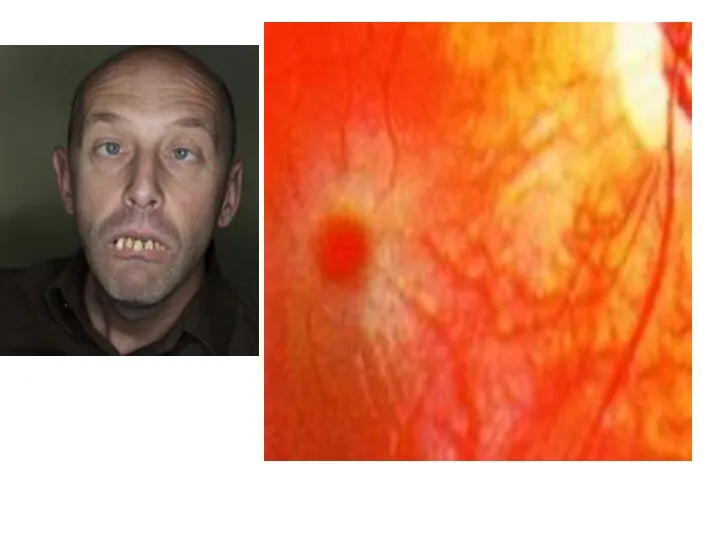
Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация)
Аутосомно-доминантный тип наследования
Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация)
Аутосомно-доминантный тип наследования

Болезнь липидного обмена
Болезнь Тея-Сакса. Ганглиозидоз. Наследуются по аутосомно-рецессивному типу. Ганглиозиды —
Болезнь липидного обмена
Болезнь Тея-Сакса. Ганглиозидоз. Наследуются по аутосомно-рецессивному типу. Ганглиозиды —

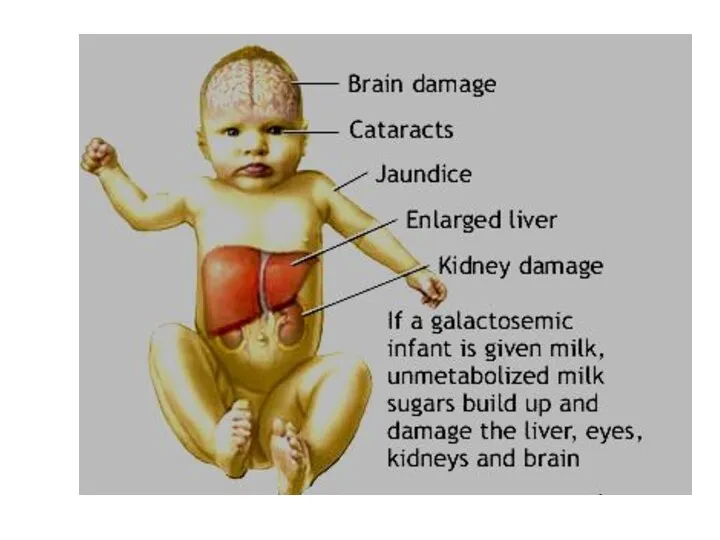
Болезнь углеводного обмена
Галактоземия. Заболевание наследуется по аутосомно-рецессивному типу.
Галактоза входит в
Болезнь углеводного обмена
Галактоземия. Заболевание наследуется по аутосомно-рецессивному типу.
Галактоза входит в

Болезни аминокислотного обмена
Фенилкетонурия
(фенилпировиноградная олигофрения). Заболевание, обусловлено дефектом фермента фенилаланингидроксилазы. Нарушается
Болезни аминокислотного обмена
Фенилкетонурия
(фенилпировиноградная олигофрения). Заболевание, обусловлено дефектом фермента фенилаланингидроксилазы. Нарушается

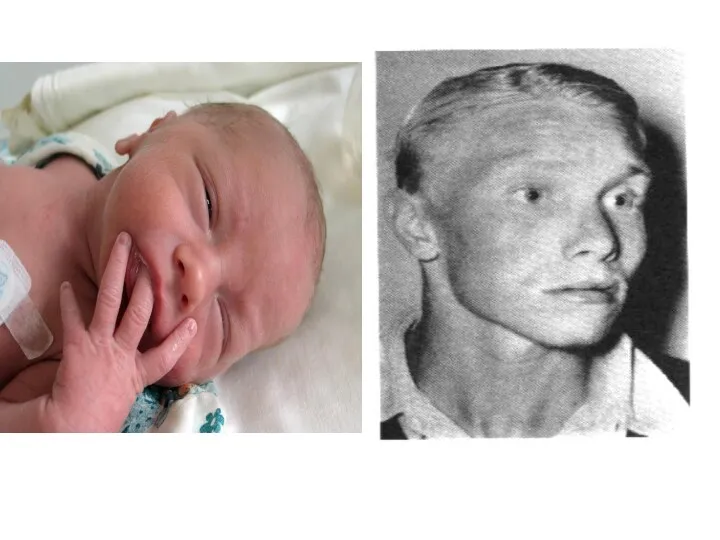
Фенилкетонурия
Клинические признаки: повышенная возбудимость и тонус мышц, тремор, «мышиный» запах, умственная
Фенилкетонурия
Клинические признаки: повышенная возбудимость и тонус мышц, тремор, «мышиный» запах, умственная

Альбинизм
При этом заболевании в коже не образуется меланин. Первичным биохимическим дефектом
Альбинизм
При этом заболевании в коже не образуется меланин. Первичным биохимическим дефектом



Самая большая в мире семья альбиносов насчитывает 10 человек.
В довольно необычной
Самая большая в мире семья альбиносов насчитывает 10 человек.
В довольно необычной


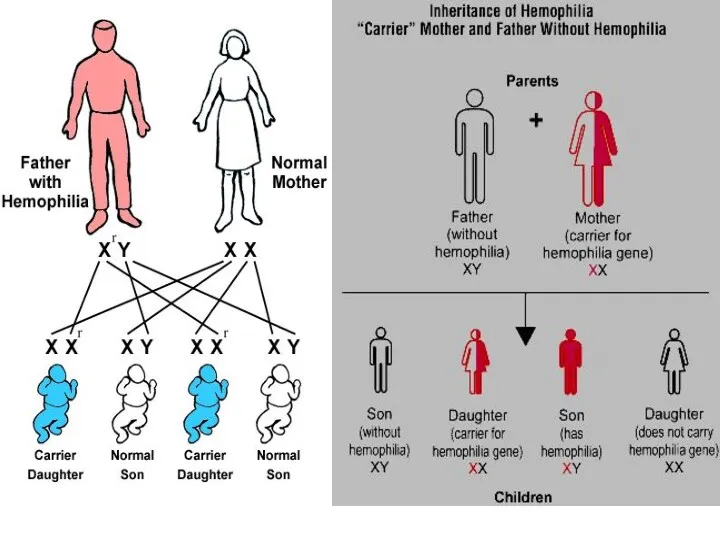
Гемофилия А - тяжелое заболевание, обусловленное дефектом фактора VIII свертывания крови.
Гемофилия А - тяжелое заболевание, обусловленное дефектом фактора VIII свертывания крови.
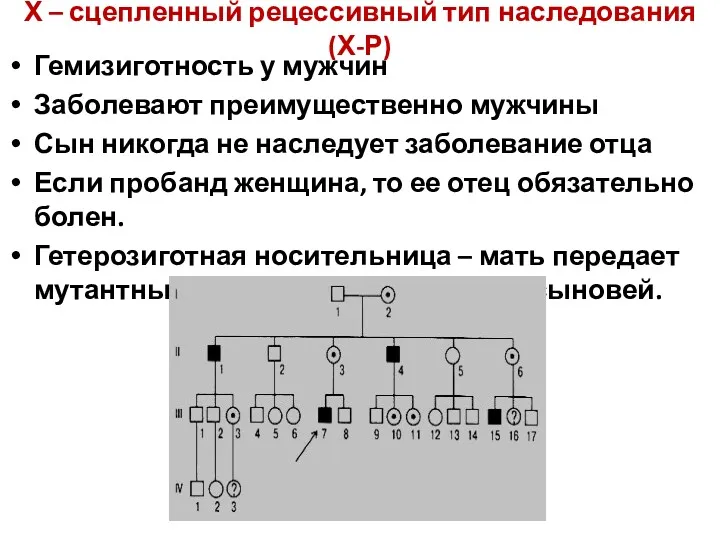
Х – сцепленный рецессивный тип наследования (Х-Р)
Гемизиготность у мужчин
Заболевают преимущественно мужчины
Сын
Х – сцепленный рецессивный тип наследования (Х-Р)
Гемизиготность у мужчин
Заболевают преимущественно мужчины
Сын

Дальтонизм
Дальтонизм

Гипофосфатемия
(витамин D-резистентная форма рахита)
Гипофосфатемия
(витамин D-резистентная форма рахита)

Х – сцепленный доминантный тип наследования (Х-Д)
У здоровых родителей все дети
Х – сцепленный доминантный тип наследования (Х-Д)
У здоровых родителей все дети

Y – сцепленный тип наследования
Голандрический тип наследования
Передача только по мужской
Y – сцепленный тип наследования
Голандрический тип наследования
Передача только по мужской
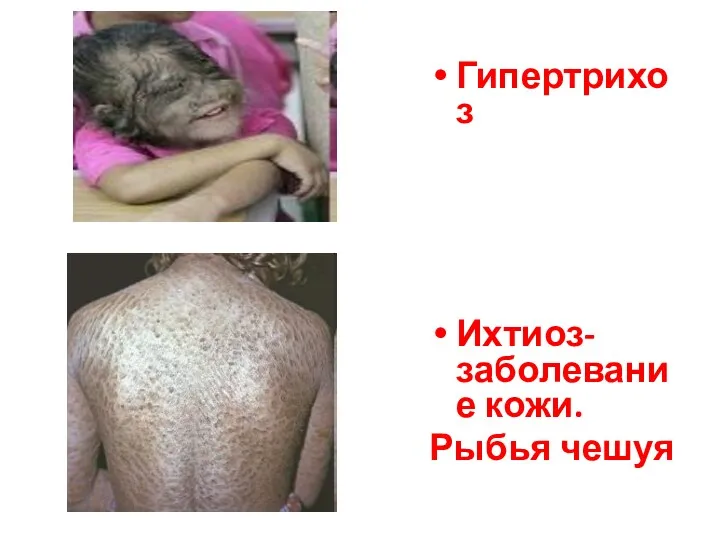
Гипертрихоз
Ихтиоз- заболевание кожи.
Рыбья чешуя
Гипертрихоз
Ихтиоз- заболевание кожи.
Рыбья чешуя



МУЛЬТИФАКТОРИАЛЬНЫЕ БОЛЕЗНИ
С наследственной предрасположен-ностью развиваются у людей с
МУЛЬТИФАКТОРИАЛЬНЫЕ БОЛЕЗНИ
С наследственной предрасположен-ностью развиваются у людей с
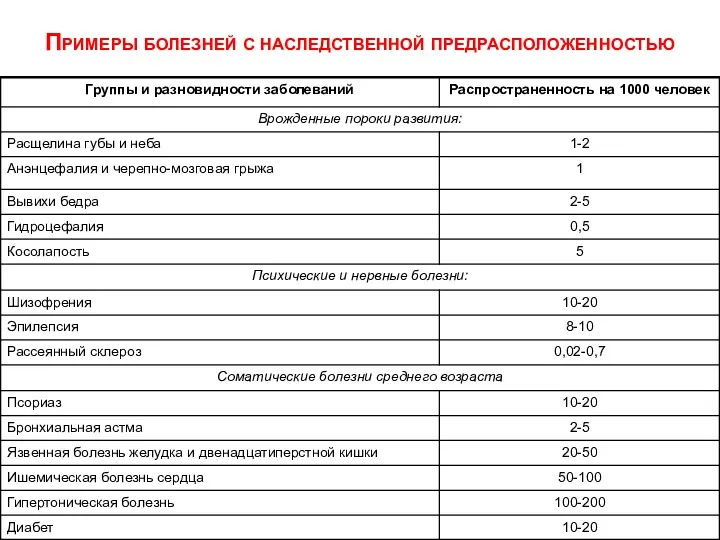
Примеры болезней с наследственной предрасположенностью
Примеры болезней с наследственной предрасположенностью

ШИЗОФРЕНИЯ
ШИЗОФРЕНИЯ

ХРОMOСОМНЫE БОЛЕЗНИ
ХРОMOСОМНЫE БОЛЕЗНИ

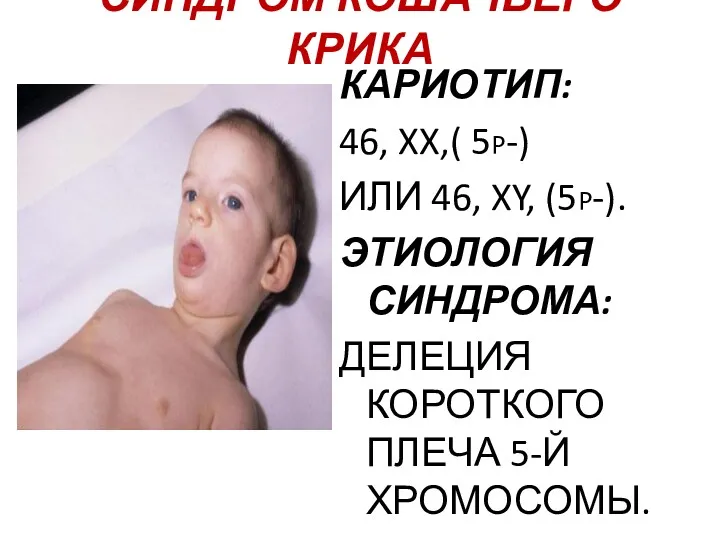
СИНДРОМ КОШАЧЬЕГО КРИКА
КАРИОТИП:
46, XX,( 5p-)
ИЛИ 46, XY, (5p-).
СИНДРОМ КОШАЧЬЕГО КРИКА
КАРИОТИП:
46, XX,( 5p-)
ИЛИ 46, XY, (5p-).
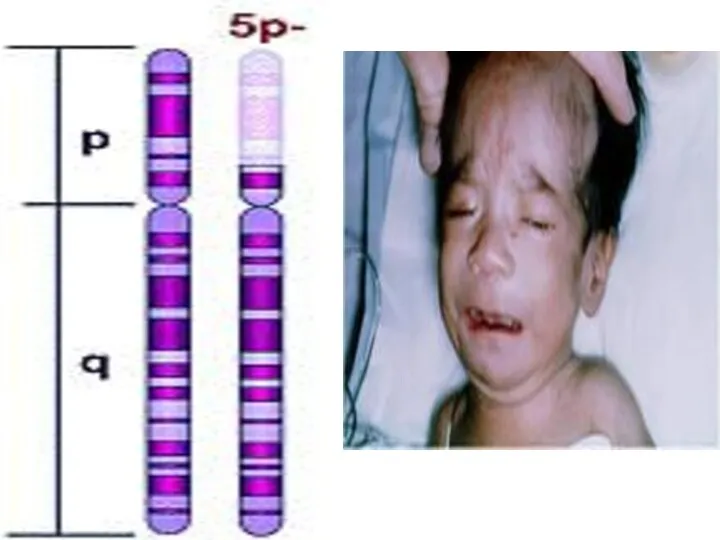
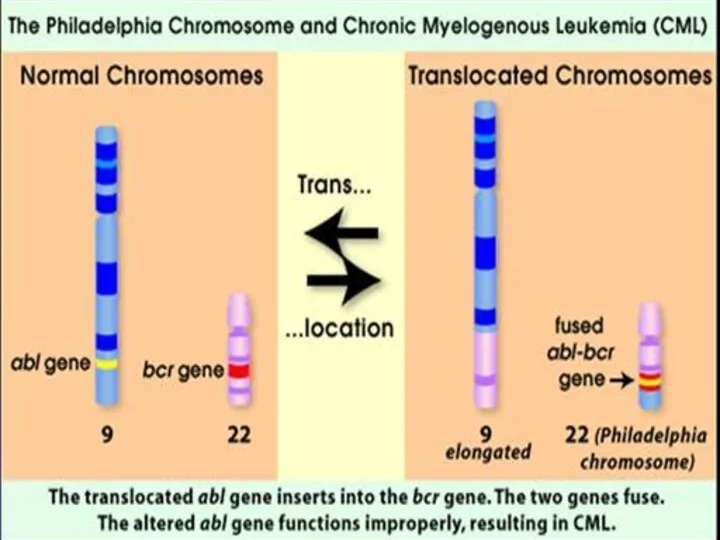
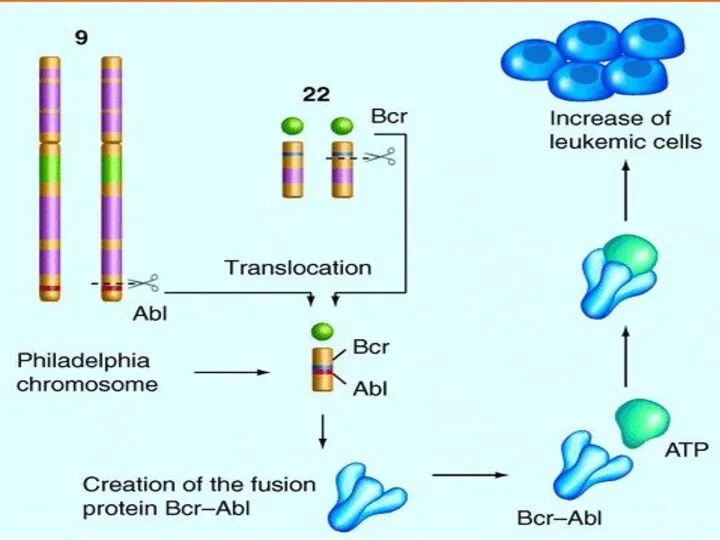

ФРАГИЛЬНАЯ Х-ХРОМОСОМА
ФРАГИЛЬНАЯ Х-ХРОМОСОМА
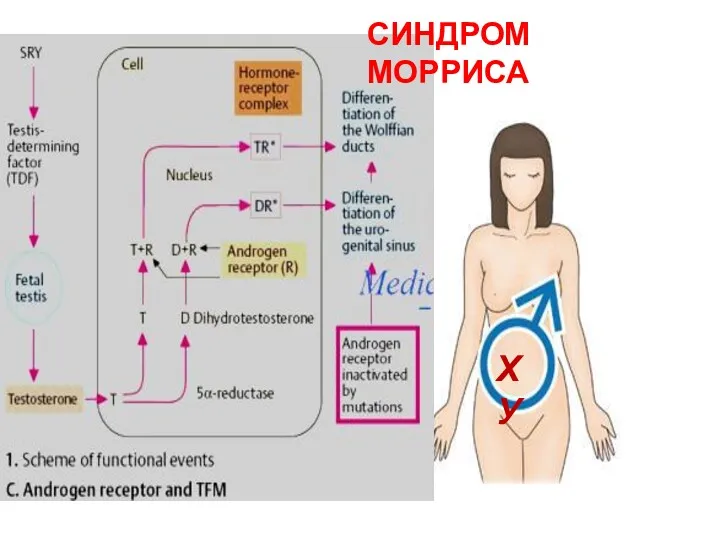
ХУ
СИНДРОМ МОРРИСА
ХУ
СИНДРОМ МОРРИСА
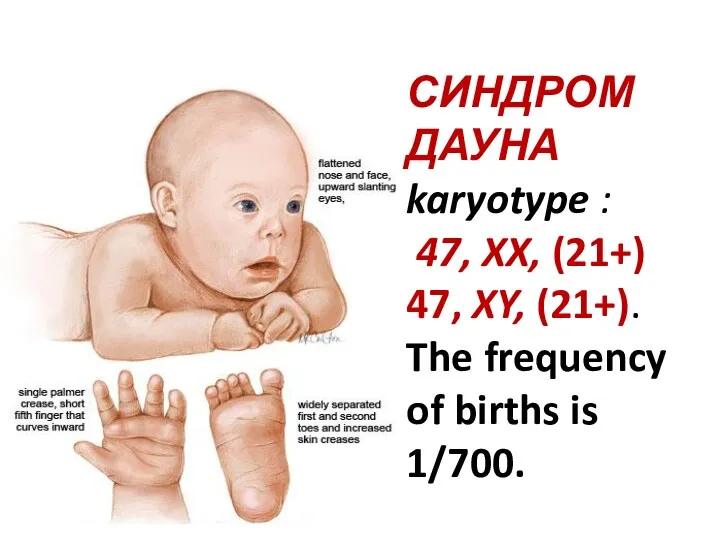
СИНДРОМ ДАУНА
karyotype :
47, XX, (21+)
47, XY, (21+). The frequency
СИНДРОМ ДАУНА
karyotype :
47, XX, (21+)
47, XY, (21+). The frequency

Синдром Дауна
Синдром - это комплекс симптомов, характерных для определенного заболевания. Синдром
Синдром Дауна
Синдром - это комплекс симптомов, характерных для определенного заболевания. Синдром
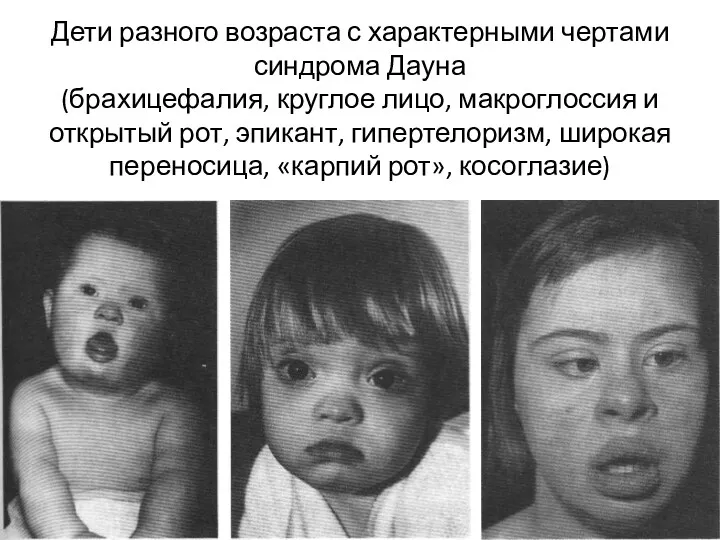
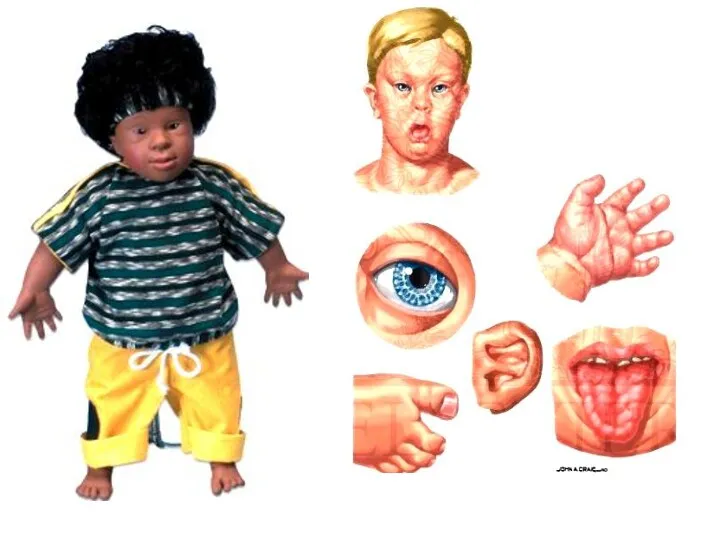
Дети разного возраста с характерными чертами синдрома Дауна
(брахицефалия, круглое лицо,
Дети разного возраста с характерными чертами синдрома Дауна (брахицефалия, круглое лицо,
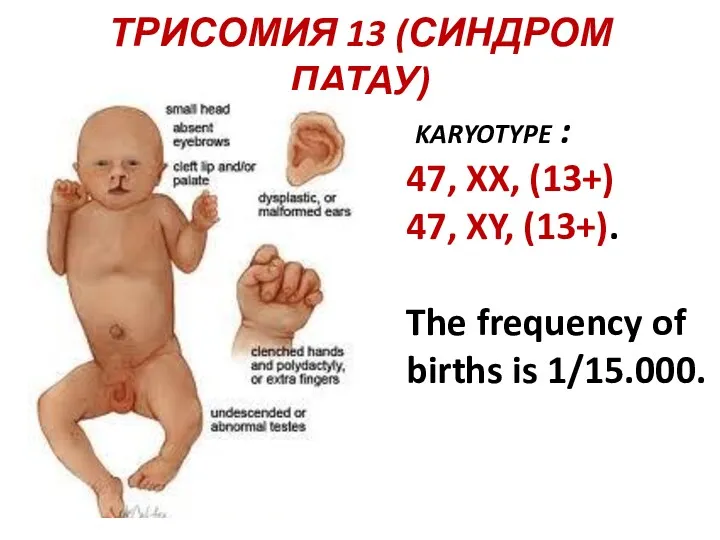
ТРИСОМИЯ 13 (СИНДРОМ ПАТАУ)
karyotype :
47, XX, (13+)
47, XY,
ТРИСОМИЯ 13 (СИНДРОМ ПАТАУ)
karyotype :
47, XX, (13+)
47, XY,
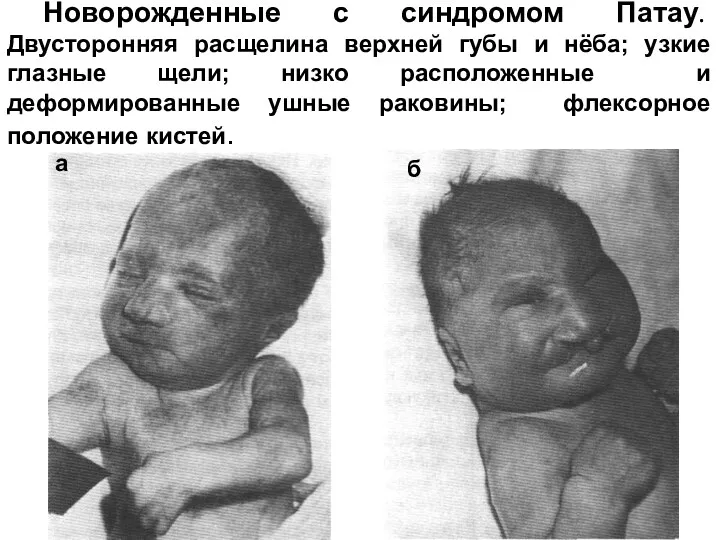
Новорожденные с синдромом Патау. Двусторонняя расщелина верхней губы и нёба; узкие
Новорожденные с синдромом Патау. Двусторонняя расщелина верхней губы и нёба; узкие
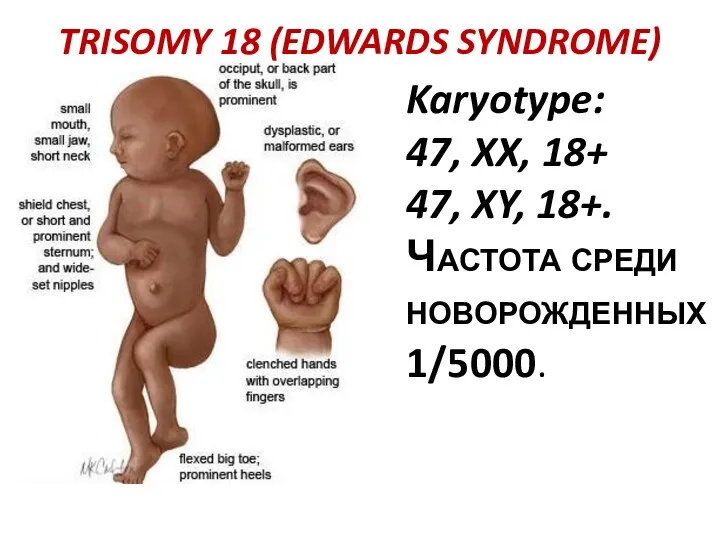
TRISOMY 18 (EDWARDS SYNDROME)
Karyotype:
47, XX, 18+
47, XY, 18+.
Частота
TRISOMY 18 (EDWARDS SYNDROME)
Karyotype:
47, XX, 18+
47, XY, 18+.
Частота


Синдром Эдвардса (трисомия 18).
Смерть до 2-3 месяцев.
Фенотип: узкий лоб,
Синдром Эдвардса (трисомия 18).
Смерть до 2-3 месяцев.
Фенотип: узкий лоб,
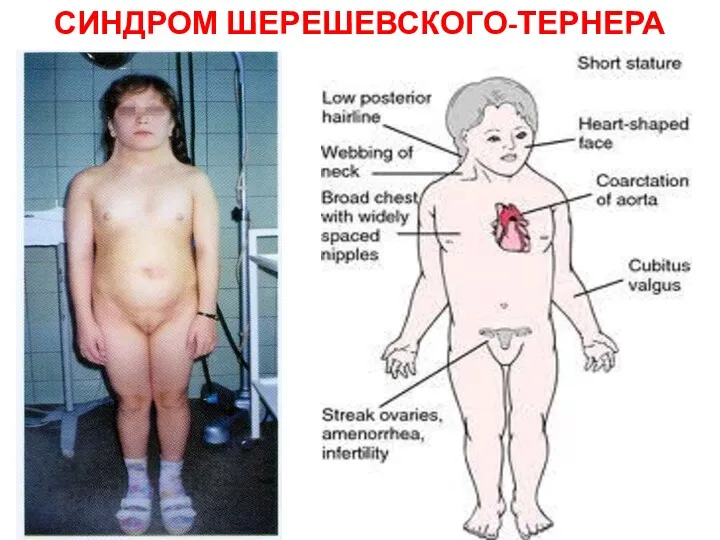
СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА
СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА
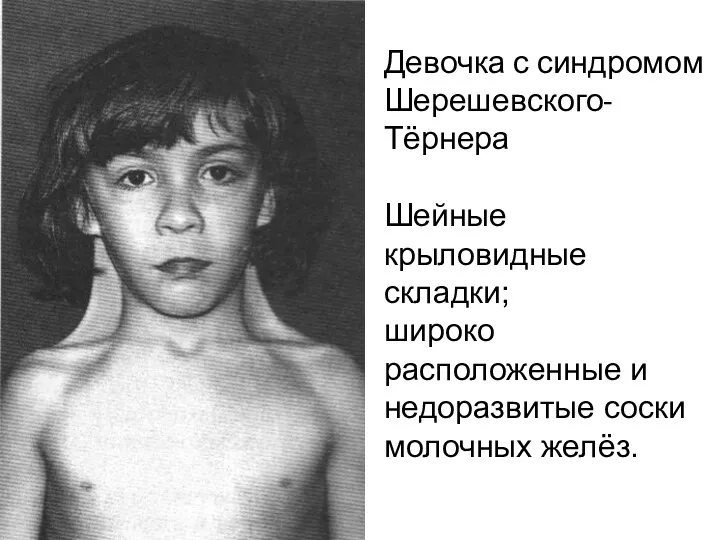
Девочка с синдромом Шерешевского-Тёрнера
Шейные крыловидные складки;
широко расположенные и недоразвитые соски
Девочка с синдромом Шерешевского-Тёрнера Шейные крыловидные складки; широко расположенные и недоразвитые соски

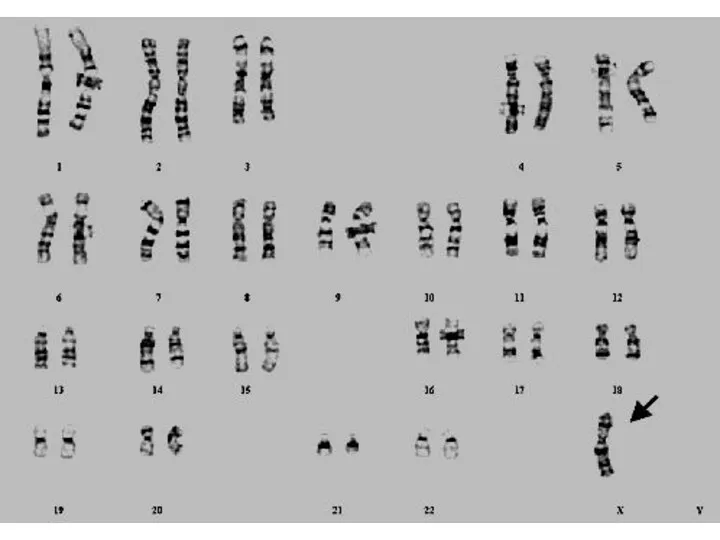
Синдром Шерешевского-Тернера
Причиной синдрома является отсутствие одной из Х-хромосом в женском организме.
Синдром Шерешевского-Тернера
Причиной синдрома является отсутствие одной из Х-хромосом в женском организме.
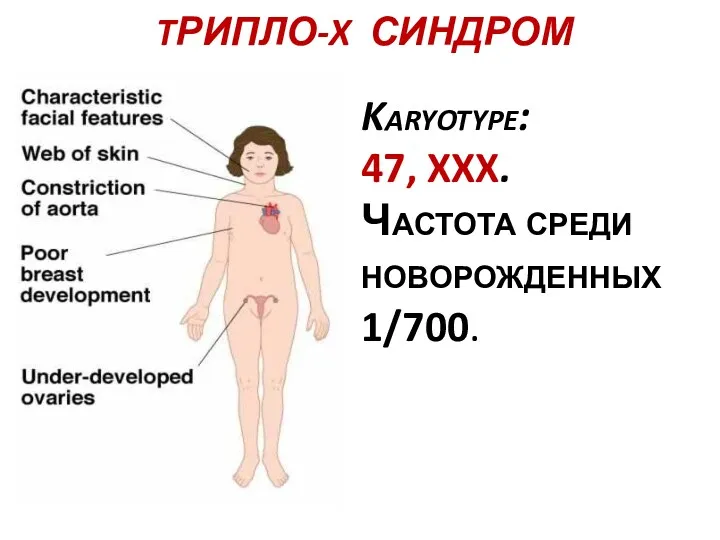
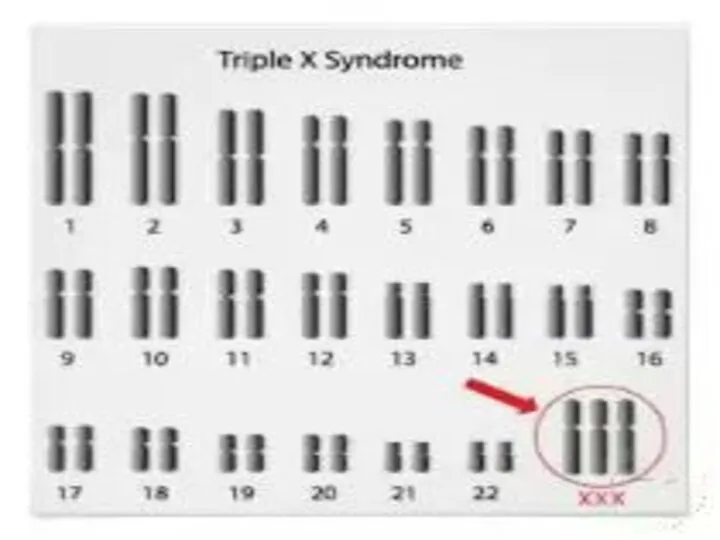
TРИПЛО-X СИНДРОМ
Karyotype:
47, XXX.
Частота среди новорожденных 1/700.
TРИПЛО-X СИНДРОМ
Karyotype:
47, XXX.
Частота среди новорожденных 1/700.

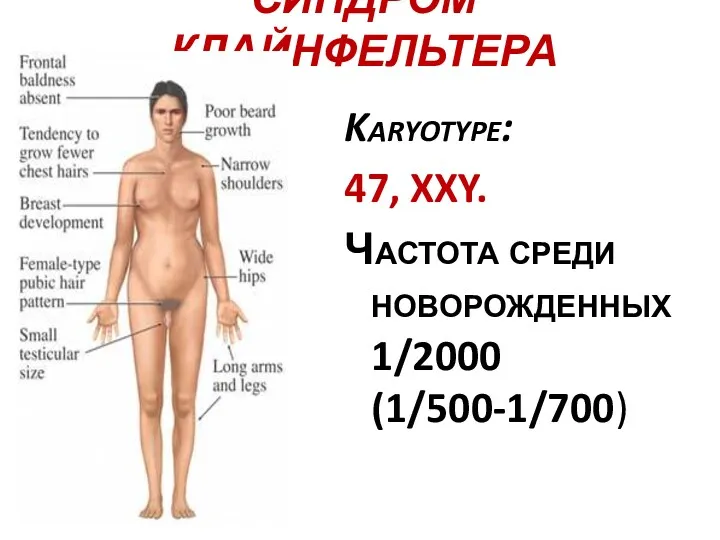
СИНДРОМ КЛАЙНФЕЛЬТЕРА
Karyotype:
47, XXY.
Частота среди новорожденных 1/2000 (1/500-1/700)
СИНДРОМ КЛАЙНФЕЛЬТЕРА
Karyotype:
47, XXY.
Частота среди новорожденных 1/2000 (1/500-1/700)

Синдром Клайнфельтера
Синдром Клайнфельтера был описан в 1942 г. Причиной синдрома является
Синдром Клайнфельтера
Синдром Клайнфельтера был описан в 1942 г. Причиной синдрома является

Синдром Клайнфельтера
Высокий рост, гинекомастия, женский тип оволосения на лобке.
Синдром Клайнфельтера
Высокий рост, гинекомастия, женский тип оволосения на лобке.

СИНДРОМ XYY (полисомияY)
karyotype :
47, XYY.
Частота среди новорожденных 1/2.000.
СИНДРОМ XYY (полисомияY)
karyotype :
47, XYY.
Частота среди новорожденных 1/2.000.

Полисомия Y
у мужчин
(синдром «супермужчины»)
Полисомия Y
у мужчин
(синдром «супермужчины»)

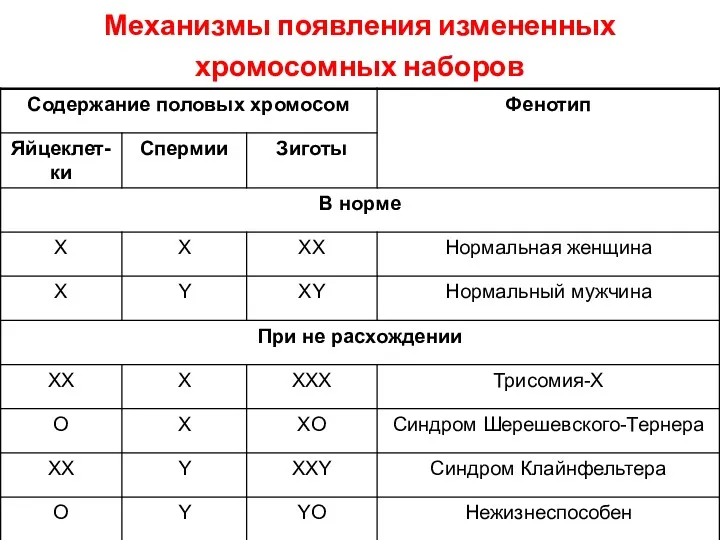
Механизмы появления измененных хромосомных наборов
Механизмы появления измененных хромосомных наборов
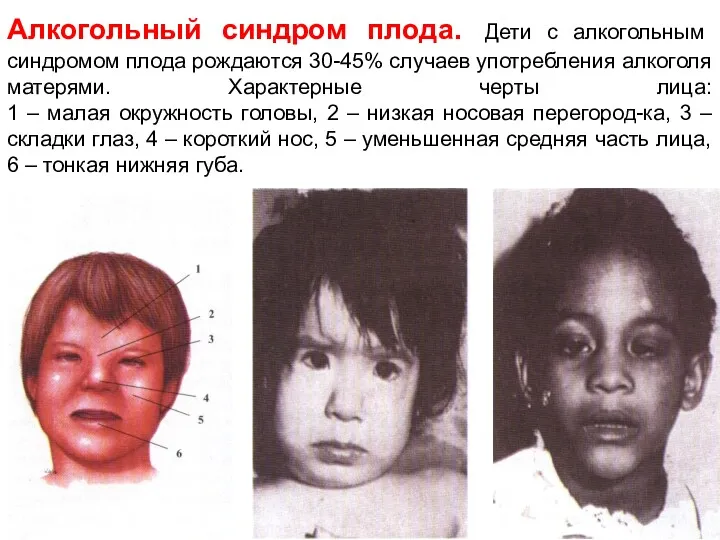
Алкогольный синдром плода. Дети с алкогольным синдромом плода рождаются 30-45% случаев
Алкогольный синдром плода. Дети с алкогольным синдромом плода рождаются 30-45% случаев

Генетические болезни соматических клеток
Генетические болезни соматических клеток выделены в отдельную группу
Генетические болезни соматических клеток
Генетические болезни соматических клеток выделены в отдельную группу

Рак - как наследственное заболевание
Среди многочисленных мульти-факториальных болезней большую
Рак - как наследственное заболевание
Среди многочисленных мульти-факториальных болезней большую

Митохондриальные болезни
Митохондриальные болезни — разнообразная группа заболеваний, обусловленных дефектами митохондриального
Митохондриальные болезни
Митохондриальные болезни — разнообразная группа заболеваний, обусловленных дефектами митохондриального

Экологическая генетика
Экологическая генетика человека изучает влияние факторов среды обитания на наследственность
Экологическая генетика
Экологическая генетика человека изучает влияние факторов среды обитания на наследственность
 Общая пропедевтика мочевыделительной системы
Общая пропедевтика мочевыделительной системы Почки и надпочечники
Почки и надпочечники Penicillin is an antibiotic
Penicillin is an antibiotic Бронхиальная астма
Бронхиальная астма Постхолецистэктомический синдром
Постхолецистэктомический синдром Владимир Петрович Филатов 1875 – 1956
Владимир Петрович Филатов 1875 – 1956 Косметика и гигиена
Косметика и гигиена Хронический гнойный средний отит
Хронический гнойный средний отит Влияние алкоголя на женский организм
Влияние алкоголя на женский организм Тубулопатия. Определение. Этиология
Тубулопатия. Определение. Этиология Қан
Қан Психологическая профилактика алкогольной зависимости у медработников в условиях медицинского учреждения
Психологическая профилактика алкогольной зависимости у медработников в условиях медицинского учреждения Принципы диагностики синдрома мальарбсорбции у детей
Принципы диагностики синдрома мальарбсорбции у детей Реконструкция проксимального отдела бедра после неудач в лечении травм
Реконструкция проксимального отдела бедра после неудач в лечении травм Клинический разбор
Клинический разбор Создание новой модели медицинской организации, оказывающей первичную медико-санитарную помощь
Создание новой модели медицинской организации, оказывающей первичную медико-санитарную помощь Бронхиальная астма, что нового?
Бронхиальная астма, что нового? Санитарно-эпидемиологическая характеристика отходов
Санитарно-эпидемиологическая характеристика отходов Съемные ортодонтические аппараты различного типа действия
Съемные ортодонтические аппараты различного типа действия Современные методы, позволяющие повысить эффективность имплантационных технологий
Современные методы, позволяющие повысить эффективность имплантационных технологий Предмет и задачи патологии. Нозология
Предмет и задачи патологии. Нозология Синдром слабости синусового узла
Синдром слабости синусового узла Реализация регионального проекта Укрепление общественного здоровья
Реализация регионального проекта Укрепление общественного здоровья Нерв тіні
Нерв тіні Патофизиология ожоговой болезни. Интенсивная терапия ожоговой болезни и ожогового шока у детей
Патофизиология ожоговой болезни. Интенсивная терапия ожоговой болезни и ожогового шока у детей Патогенез воспалительных заболеваний пародонта
Патогенез воспалительных заболеваний пародонта Сурфактантная терапия
Сурфактантная терапия Фармакология как наука и учебная дисциплина
Фармакология как наука и учебная дисциплина