- Груз наследственных болезней в популяциях человека

Содержание
- 2. Регистр врожденной патологии частота аутосомно-доминантных заболеваний составляет 1395,4 на 1 млн. новорожденных и составляет 0,14%; аутосомно-рецессивных
- 3. Регистр является лучшим в мире источником информации о частоте наследственных болезней в популяциях человека, хотя и
- 4. Собственно наследственная патология, которая включает менделирующую (аутосомно-доминантную, аутосомно-рецессивную, Х-сцепленную рецессивную и хромосомную) патологию, встречается примерно у
- 5. Данные о частоте наследственных болезней в популяции Британской Колумбии – не единственные в мировой литературе, впервые
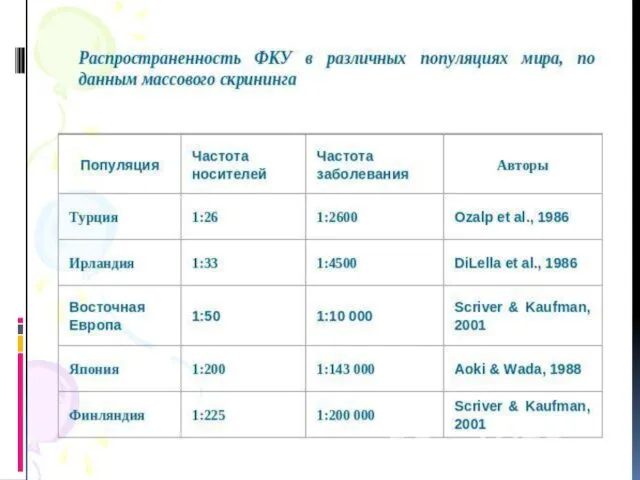
- 6. Частота основных типов наследственных болезней на 1000 новорожденных по данным ряда источников
- 7. Согласно данным, представленным в этой таблице, наибольшим колебаниям подвержена оценка частоты аутосомно-доминантных болезней, которая, по данным
- 8. Пенетрантность этих признаков в терминах патологических состояний достаточно низкая, поэтому их включение в список регистрируемых доминантных
- 9. Дифференциация медицинской генетики на отдельные дисциплины
- 10. ОБЩИЕ ПОЛОЖЕНИЯ, ТИПЫ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
- 11. Наследственные заболевания человека - патологические состояния, причиной которых является изменение генетического материала.
- 12. Моногенные или менделирующие болезни; Хромосомные болезни; Митохондриальные болезни; Геномный импринтинг или болезни импринтинга; Мультифакторные болезни или
- 13. 1 тип - моногенные болезни
- 14. Моногенные болезни часто называют менделирующими, так как они наследуются согласно правилам Менделя.
- 15. Болезни с аутосомно-доминантным наследованием Синдром Марфана — заболевание наследственного типа, при котором поражается соединительная ткань с
- 16. Синдром Марфана Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил
- 17. Синдром Марфана
- 18. Клинические признаки синдрома Марфана — гиперподвижность суставов; — аномалии строения тазобедренного сустава; — кифоз, сколиоз; —
- 19. Болезни с аутосомно-рецессивным наследованием Фенилкетонурия – наиболее распространенное нарушение обмена аминокислот. Частота - 1 : 8000
- 21. КЛИНИЧЕСКИЕ ПРИЗНАКИ ФКУ Вялость или наоборот, повышенная двигательная активность малыша; ребенок не проявляет интерес к окружающему
- 22. Ребенок с фенилкетонурией (ФКУ)
- 23. Внутриутробная диагностика фенилкетонурии невозможна, так как существуют определенные сложности при получении биоматериала ребенка для проведения исследования.
- 24. Генетический дефект может быть вызван: хроническим алкоголизмом одного или обоих родителей, длительным влиянием радиоактивных волн на
- 25. Диагностируют ФКУ, опираясь на совокупность генеалогических данных, результаты биохимических и клинических исследований: возможно мама и папа
- 26. Скрининг на ФКУ может проводится при помощи микробиологического теста Гатри - основан на принципе молекулярного антагонизма,
- 27. Забор крови производится у новорожденных в возрасте 4–5 дней.
- 29. Основу лечения ФКУ составляет диетотерапия, при которой должно строго ограничиваться поступление в организм белка с продуктами
- 30. Что можно, что нельзя Продукты, назначаемые для лечения фенилкетонурии: Гидролизаты белковые, Смеси L-аминокислотные, не содержащие фенилаланин,
- 31. 2. Хромосомные болезни Второй тип наследственных болезней – хромосомные, так как изменения затрагивают хромосомы, в них
- 32. Хромосомные аутосомные болезни Синдром Дауна Синдром Патау
- 33. Хромосомные аутосомные болезни Синдром Эдвардса
- 34. Хромосомные гоносомные болезни Синдром Клайнфельтера Синдром Шерешевского-Тернера
- 35. Делеционные синдромы Синдром Вольфа-Хиршхорна
- 36. Делеционные синдромы Синдром кошачьего крика
- 37. НАСЛЕДОВАНИЕ, СЦЕПЛЕННОЕ С Х-ХРОМОСОМОЙ Миопатия Дюшена
- 38. Миопатия Дюшена Миопатия Дюшена - это первичная мышечная дистрофия, наследственное дегенеративное заболевание, в основе которого лежит
- 39. Миопатия Дюшена
- 40. Гемофилия
- 41. Гемофилия Гемофилия — это наследственное заболевание, связанное с нарушением функции свертывания крови в результате мутации в
- 42. Гемофилия
- 43. Гемофилия типа A возникает в результате недостаточности фактора свертывания VIII (антигемофильный глобулин- белковая молекула, один из
- 44. Гемофилии типов А и В. Мутация в хромосоме Х. По правилам наследования гена, сцепленного с Х-хромосомой:
- 45. Синдром ломкой Х-хромосомы
- 46. Синдром ломкой Х-хромосомы (ОMIM №309550) — Х-сцепленное заболевание с задержкой умственного развития, вызванное мутациями в гене
- 47. Продукт гена FMR1, FMRP, экспрессируется во многих типах клеток, но наиболее сильно в нейронах. FMRP может
- 48. Почти все мужчины и 40-50% женщин, унаследовавших полную мутацию, будут иметь синдром ломкой Х-хромосомы. Тяжесть фенотипа
- 49. Синдром ломкой Х-хромосомы вызывает умеренную умственную отсталость у мужчин и легкую умственную задержку у женщин. Наиболее
- 50. К настоящему времени никакого патогенетического лечения при синдроме ломкой Х-хромосомы нет. Помощь направлена на обучение и
- 51. 3. Митохондриальные болезни митохондриальные болезни, возникают в том случае, если мутации затрагивают митохондриальную ДНК. Примером митохондриальных
- 52. Термин «митохондриальные болезни» вошел в медицинский научный лексикон лишь несколько лет назад. Им обозначается много заболеваний
- 53. Каждая клетка содержит сотни, а иногда и тысячи митохондрий. Основная, но далеко не единственная функция этих
- 54. Рис. Электронно-микроскопическая Рис. Схема строения митохондрии. фотография митохондрии человека в Сложная структура в поперечном разрезе митохондрии
- 55. Так как мтДНК содержится в цитоплазме клеток, она наследуется только по материнской линии. В цитоплазме яйцеклеток
- 56. Как правило, все типы мтДНК идентичны, такое состояние носит название гомоплизии. Появление мутации в одной из
- 57. Учитывая, что энергетические потребности разных тканей организма различны, а наиболее энергопотребляющей является нервная система, то вполне
- 60. Классификация митохондриальных болезней К 1 классу относятся первичные дефекты окислительного фосфорилирования. Ко 2 классу относятся МТ
- 61. Атрофия зрительных нервов Лебера обусловлена мутациями в генах мтДНК, кодирующих субъединицы комплекса 1. Наиболее частая мутация
- 62. Синдром Лея (подострая некротизирующая энцефаломиелопатия) также относится к этому классу. . Синдром Лея часто ассоциируется с
- 63. Синдром Лея, Подострая некротизирующая энцефаломиопатия — редкий наследственный нейрометаболический синдром, поражающий ЦНС. В основном заболевают дети
- 64. Синдром нейропатии, атаксии и пигментной дистрофии сетчатки (NARP) может проявляться как в младенчестве, так и позже,
- 65. Синдром нейропатии, атаксии и пигментной дистрофии сетчатки (NARP)
- 66. Синдром миоклонус-эпилепсии и рваных красных мышечных волокон (MERRF), который проявляется эпилепсией, деменцией, атаксией и миопатией, возникает
- 67. Синдром миоклонус-эпилепсии и рваных красных мышечных волокон (MERRF)
- 68. Синдром митохондриальной энцефаломиопатии и инсультоподобных эпизодов (MELAS)
- 69. К МТБ, обусловленным делециями или дупликациями, относятся синдром Кернса-Сайра (миопатия, мозжечковые нарушения и сердечная недостаточность) (фото)
- 70. 4.Болезни геномного импринтинга геномный импринтинг и болезни импринтинга, это класс болезней, которые не соответствуют менделевскому наследованию,
- 71. Болезни импринтинга (однородительская дисомия)
- 72. Синдром Х-ломкой хромосомы
- 73. Геномный импринтинг – эпигенетическое явление, при котором наследуются изменения генной активности, обусловленные родительским происхождением хромосом или
- 74. Первоначальный «отпечаток», созданный в половых клетках, служит основанием для дальнейших модификаций в результате взаимодействий между родительскими
- 75. У человека эффект импринтинга обнаружен в связи с наличием в хромосомном наборе фрагментов или целых хромосом
- 76. Регион синдрома Прадера-Вилли активен на отцовской хромосоме (при наличии делеции на отцовской хромосоме или материнской однородительской
- 78. 5. Мультифакторные болезни мультифакторные болезни или болезни с наследственной предрасположенностью, так как их формирование обусловлено взаимодействием
- 79. ПРИЧИНЫ БОЛЕЗНЕЙ С НАСЛЕДСТВЕННОЙ ПРЕДРАСПОЛОЖЕННОСТЬЮ ПРИЧИНЫ СРЕДОВЫЕ СЕМЕЙНЫЕ ПОПУЛЯЦИОН-НЫЕ ГЕНЕТИЧЕСКИЕ ГЕНЫ ПРЕДРАСПО-ЛОЖЕННОСТИ СТОХАСТИЧЕСКИЕ (случайные) ГЕНЕТИЧЕСКИЙ ФОН
- 80. К ним относятся все хронические неинфекционные заболевания, такие как диабет, атеросклероз, бронхиальная астма и др., а
- 81. Одним из способов выявления генов, которые могут иметь отношение к предрасположенности к МЗ, является изучение ассоциаций
- 82. 6. Заболевания, обусловленные преимущественно действием внешнесредовых факторов – это травмы, частые инфекционные заболевания.
- 83. Влияние курения на легкие человека
- 84. Ожоги
- 86. Скачать презентацию

Регистр врожденной патологии
частота аутосомно-доминантных заболеваний составляет 1395,4 на 1 млн. новорожденных
Регистр врожденной патологии
частота аутосомно-доминантных заболеваний составляет 1395,4 на 1 млн. новорожденных

Регистр является лучшим в мире источником информации о частоте наследственных болезней
Регистр является лучшим в мире источником информации о частоте наследственных болезней

Собственно наследственная патология, которая включает менделирующую (аутосомно-доминантную, аутосомно-рецессивную, Х-сцепленную рецессивную и
Собственно наследственная патология, которая включает менделирующую (аутосомно-доминантную, аутосомно-рецессивную, Х-сцепленную рецессивную и

Данные о частоте наследственных болезней в популяции Британской Колумбии – не
Данные о частоте наследственных болезней в популяции Британской Колумбии – не
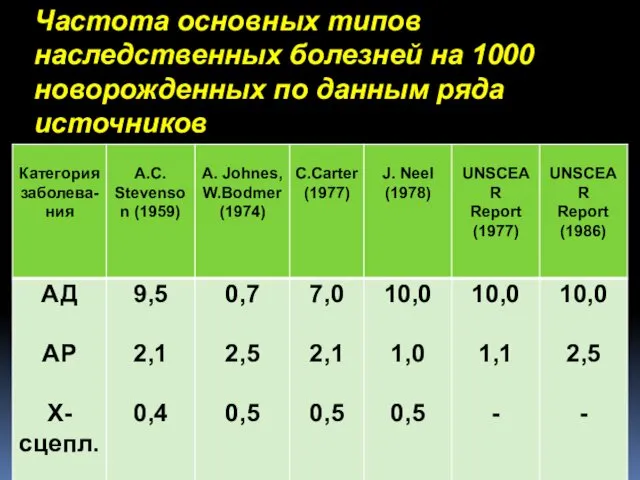
Частота основных типов наследственных болезней на 1000 новорожденных по данным ряда
Частота основных типов наследственных болезней на 1000 новорожденных по данным ряда

Согласно данным, представленным в этой таблице, наибольшим колебаниям подвержена оценка частоты
Согласно данным, представленным в этой таблице, наибольшим колебаниям подвержена оценка частоты

Пенетрантность этих признаков в терминах патологических состояний достаточно низкая, поэтому их
Пенетрантность этих признаков в терминах патологических состояний достаточно низкая, поэтому их

Дифференциация медицинской генетики на отдельные дисциплины
Дифференциация медицинской генетики на отдельные дисциплины

ОБЩИЕ ПОЛОЖЕНИЯ, ТИПЫ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
ОБЩИЕ ПОЛОЖЕНИЯ, ТИПЫ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ

Наследственные заболевания человека - патологические состояния, причиной которых является изменение генетического
Наследственные заболевания человека - патологические состояния, причиной которых является изменение генетического

Моногенные или менделирующие болезни;
Хромосомные болезни;
Митохондриальные болезни;
Геномный импринтинг или болезни импринтинга;
Мультифакторные болезни
Моногенные или менделирующие болезни;
Хромосомные болезни;
Митохондриальные болезни;
Геномный импринтинг или болезни импринтинга;
Мультифакторные болезни

1 тип - моногенные болезни
1 тип - моногенные болезни

Моногенные болезни часто называют менделирующими, так как они наследуются согласно правилам
Моногенные болезни часто называют менделирующими, так как они наследуются согласно правилам

Болезни с аутосомно-доминантным наследованием
Синдром Марфана — заболевание наследственного типа, при котором
Болезни с аутосомно-доминантным наследованием
Синдром Марфана — заболевание наследственного типа, при котором

Синдром Марфана
Синдром Марфана получил своё название от фамилии французского педиатра А.
Синдром Марфана
Синдром Марфана получил своё название от фамилии французского педиатра А.
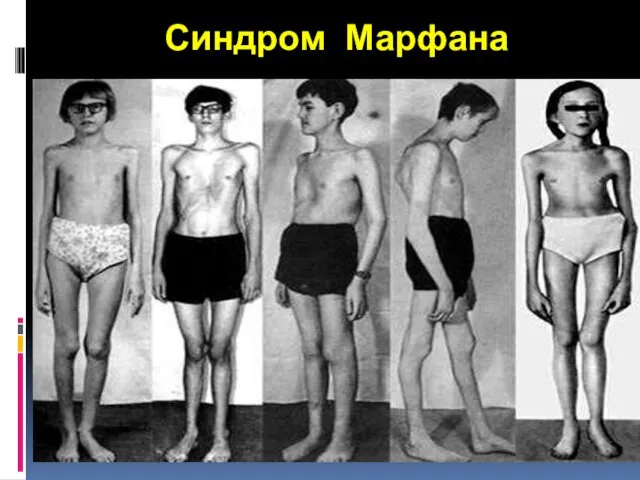
Синдром Марфана
Синдром Марфана
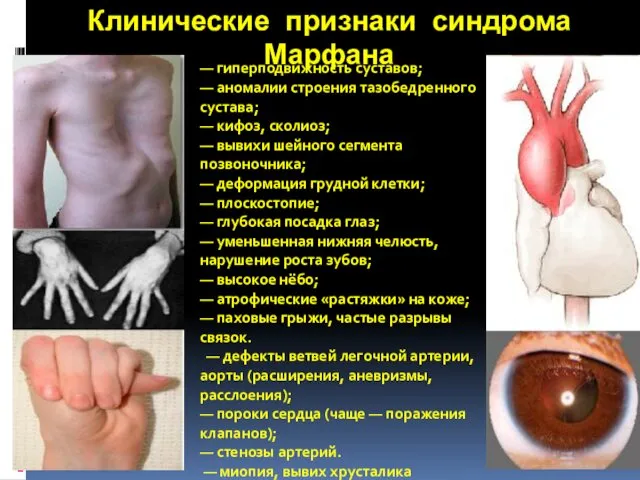
Клинические признаки синдрома Марфана
— гиперподвижность суставов;
— аномалии строения тазобедренного сустава;
— кифоз,
Клинические признаки синдрома Марфана
— гиперподвижность суставов; — аномалии строения тазобедренного сустава; — кифоз,
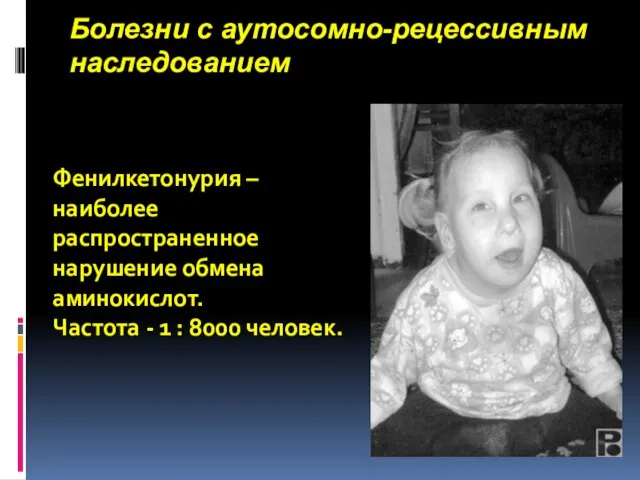
Болезни с аутосомно-рецессивным наследованием
Фенилкетонурия –
наиболее распространенное нарушение обмена аминокислот.
Частота
Болезни с аутосомно-рецессивным наследованием
Фенилкетонурия –
наиболее распространенное нарушение обмена аминокислот.
Частота

КЛИНИЧЕСКИЕ ПРИЗНАКИ ФКУ
Вялость или наоборот, повышенная двигательная активность малыша;
ребенок не
КЛИНИЧЕСКИЕ ПРИЗНАКИ ФКУ
Вялость или наоборот, повышенная двигательная активность малыша;
ребенок не

Ребенок с фенилкетонурией (ФКУ)
Ребенок с фенилкетонурией (ФКУ)

Внутриутробная диагностика фенилкетонурии невозможна, так как существуют определенные сложности при получении
Внутриутробная диагностика фенилкетонурии невозможна, так как существуют определенные сложности при получении

Генетический дефект может быть вызван:
хроническим алкоголизмом одного или обоих родителей,
Генетический дефект может быть вызван:
хроническим алкоголизмом одного или обоих родителей,

Диагностируют ФКУ, опираясь на совокупность генеалогических данных, результаты биохимических и клинических
Диагностируют ФКУ, опираясь на совокупность генеалогических данных, результаты биохимических и клинических

Скрининг на ФКУ может проводится при помощи микробиологического теста Гатри -
Скрининг на ФКУ может проводится при помощи микробиологического теста Гатри -
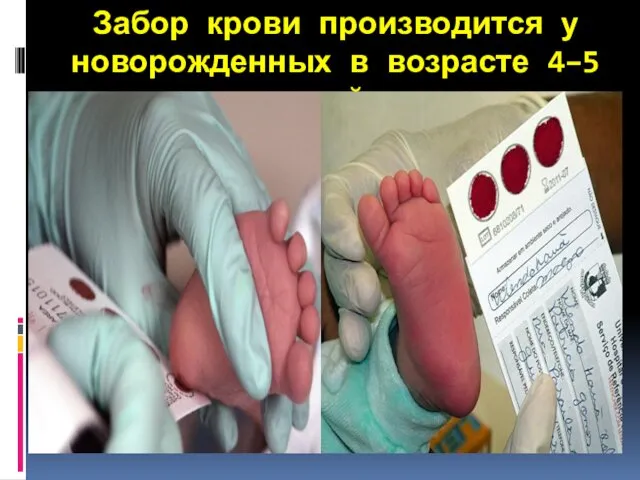
Забор крови производится у новорожденных в возрасте 4–5 дней.
Забор крови производится у новорожденных в возрасте 4–5 дней.


Основу лечения ФКУ составляет диетотерапия, при которой должно строго ограничиваться поступление
Основу лечения ФКУ составляет диетотерапия, при которой должно строго ограничиваться поступление

Что можно, что нельзя
Продукты, назначаемые для лечения фенилкетонурии:
Гидролизаты белковые,
Смеси L-аминокислотные,
Что можно, что нельзя
Продукты, назначаемые для лечения фенилкетонурии:
Гидролизаты белковые,
Смеси L-аминокислотные,

2. Хромосомные болезни
Второй тип наследственных болезней – хромосомные, так как изменения
2. Хромосомные болезни
Второй тип наследственных болезней – хромосомные, так как изменения
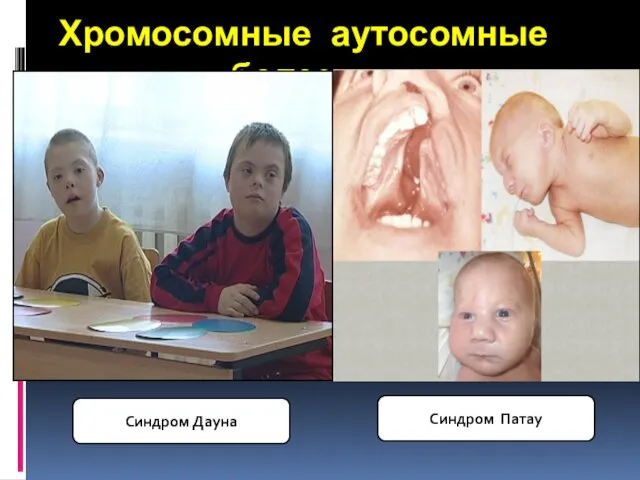
Хромосомные аутосомные болезни
Синдром Дауна
Синдром Патау
Хромосомные аутосомные болезни
Синдром Дауна
Синдром Патау

Хромосомные аутосомные болезни
Синдром Эдвардса
Хромосомные аутосомные болезни
Синдром Эдвардса
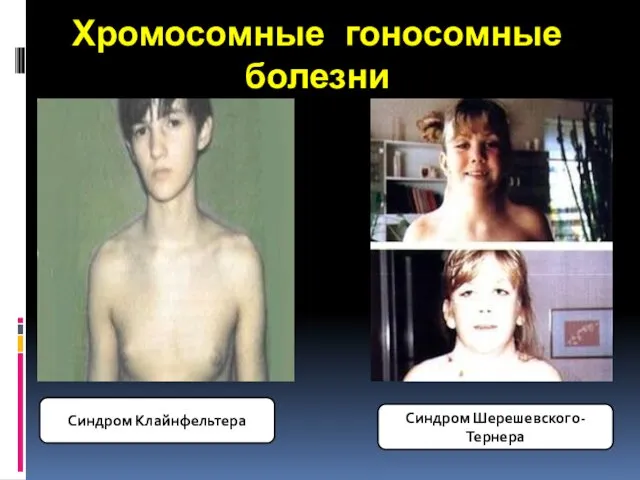
Хромосомные гоносомные болезни
Синдром Клайнфельтера
Синдром Шерешевского-Тернера
Хромосомные гоносомные болезни
Синдром Клайнфельтера
Синдром Шерешевского-Тернера
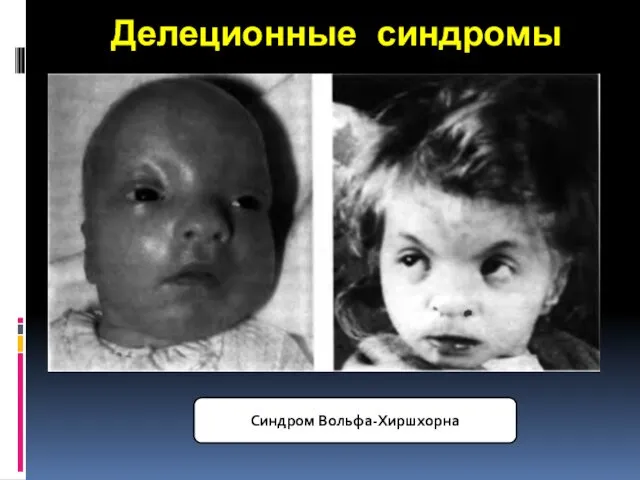
Делеционные синдромы
Синдром Вольфа-Хиршхорна
Делеционные синдромы
Синдром Вольфа-Хиршхорна

Делеционные синдромы
Синдром кошачьего крика
Делеционные синдромы
Синдром кошачьего крика
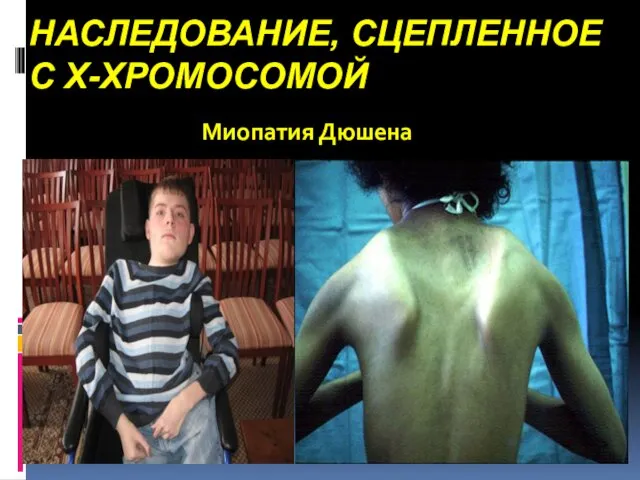
НАСЛЕДОВАНИЕ, СЦЕПЛЕННОЕ С Х-ХРОМОСОМОЙ
Миопатия Дюшена
НАСЛЕДОВАНИЕ, СЦЕПЛЕННОЕ С Х-ХРОМОСОМОЙ
Миопатия Дюшена

Миопатия Дюшена
Миопатия Дюшена - это первичная мышечная дистрофия, наследственное дегенеративное заболевание,
Миопатия Дюшена
Миопатия Дюшена - это первичная мышечная дистрофия, наследственное дегенеративное заболевание,
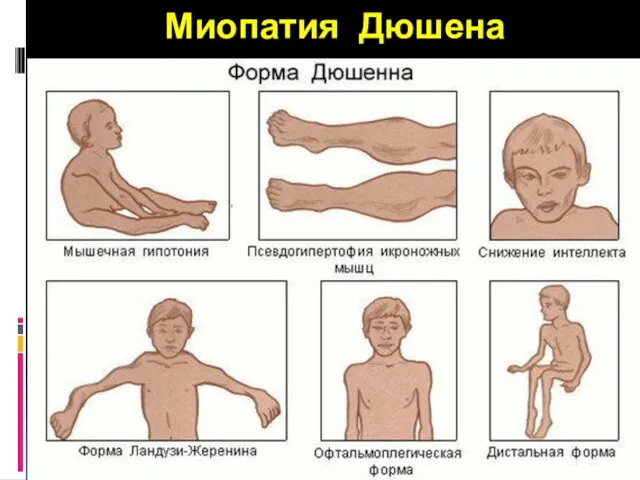
Миопатия Дюшена
Миопатия Дюшена
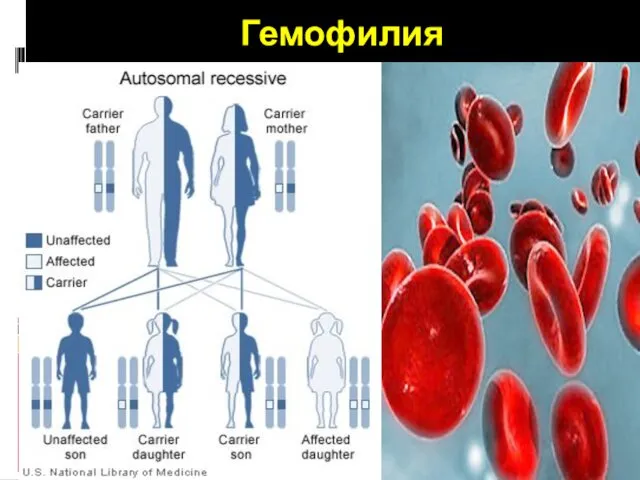
Гемофилия
Гемофилия

Гемофилия
Гемофилия — это наследственное заболевание, связанное с нарушением функции свертывания крови
Гемофилия
Гемофилия — это наследственное заболевание, связанное с нарушением функции свертывания крови
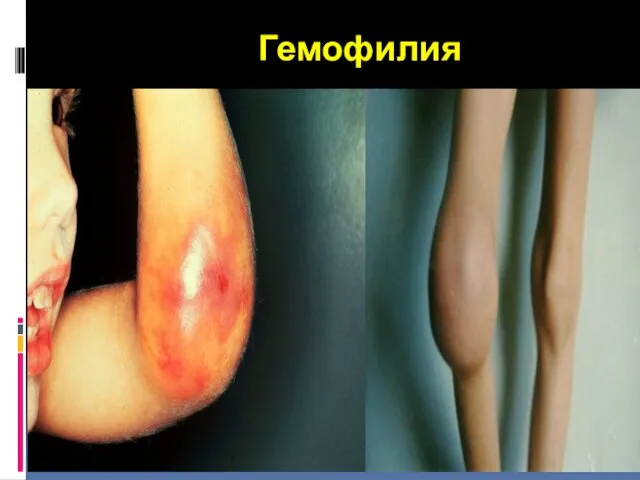
Гемофилия
Гемофилия

Гемофилия типа A возникает в результате недостаточности фактора свертывания VIII (антигемофильный глобулин-
Гемофилия типа A возникает в результате недостаточности фактора свертывания VIII (антигемофильный глобулин-

Гемофилии типов А и В.
Мутация в хромосоме Х. По правилам
Гемофилии типов А и В.
Мутация в хромосоме Х. По правилам

Синдром ломкой Х-хромосомы
Синдром ломкой Х-хромосомы

Синдром ломкой Х-хромосомы (ОMIM №309550) — Х-сцепленное заболевание с задержкой умственного
Синдром ломкой Х-хромосомы (ОMIM №309550) — Х-сцепленное заболевание с задержкой умственного

Продукт гена FMR1, FMRP, экспрессируется во многих типах клеток, но наиболее
Продукт гена FMR1, FMRP, экспрессируется во многих типах клеток, но наиболее

Почти все мужчины и 40-50% женщин, унаследовавших полную мутацию, будут иметь
Почти все мужчины и 40-50% женщин, унаследовавших полную мутацию, будут иметь

Синдром ломкой Х-хромосомы вызывает умеренную умственную отсталость у мужчин и легкую
Синдром ломкой Х-хромосомы вызывает умеренную умственную отсталость у мужчин и легкую

К настоящему времени никакого патогенетического лечения при синдроме ломкой Х-хромосомы нет.
К настоящему времени никакого патогенетического лечения при синдроме ломкой Х-хромосомы нет.

3. Митохондриальные болезни
митохондриальные болезни, возникают в том случае, если мутации затрагивают
3. Митохондриальные болезни
митохондриальные болезни, возникают в том случае, если мутации затрагивают

Термин «митохондриальные болезни» вошел в медицинский научный лексикон лишь несколько лет
Термин «митохондриальные болезни» вошел в медицинский научный лексикон лишь несколько лет

Каждая клетка содержит сотни, а иногда и тысячи митохондрий.
Основная, но
Каждая клетка содержит сотни, а иногда и тысячи митохондрий.
Основная, но
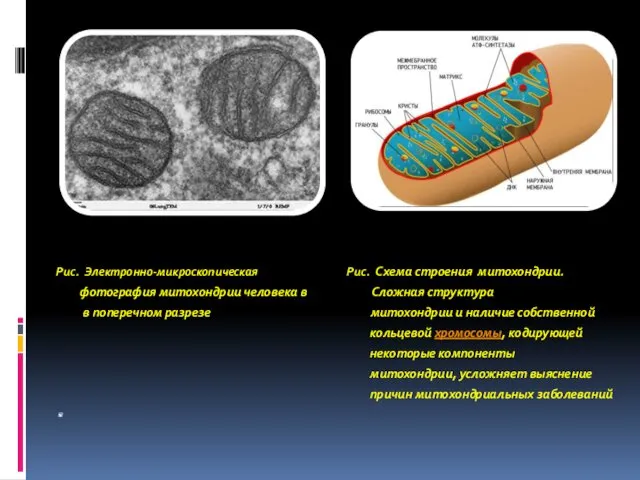
Рис. Электронно-микроскопическая Рис. Схема строения митохондрии.
фотография митохондрии человека в
Рис. Электронно-микроскопическая Рис. Схема строения митохондрии.
фотография митохондрии человека в

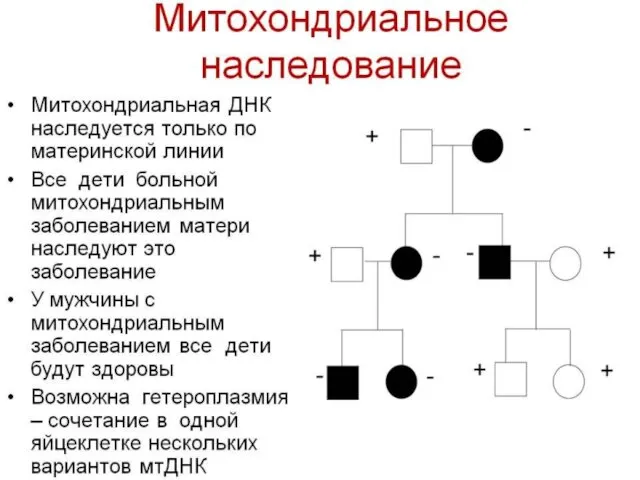
Так как мтДНК содержится в цитоплазме клеток, она наследуется только по
Так как мтДНК содержится в цитоплазме клеток, она наследуется только по

Как правило, все типы мтДНК идентичны, такое состояние носит название гомоплизии.
Как правило, все типы мтДНК идентичны, такое состояние носит название гомоплизии.

Учитывая, что энергетические потребности разных тканей организма различны, а наиболее энергопотребляющей
Учитывая, что энергетические потребности разных тканей организма различны, а наиболее энергопотребляющей
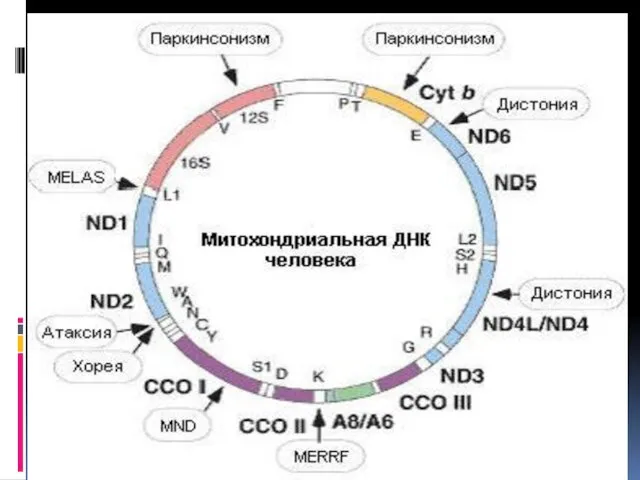

Классификация митохондриальных болезней
К 1 классу относятся первичные дефекты окислительного фосфорилирования.
Ко
Классификация митохондриальных болезней
К 1 классу относятся первичные дефекты окислительного фосфорилирования.
Ко
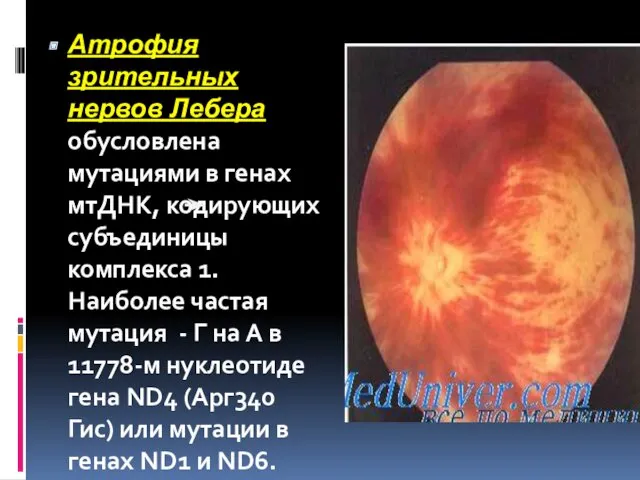
Атрофия зрительных нервов Лебера обусловлена мутациями в генах мтДНК, кодирующих субъединицы
Атрофия зрительных нервов Лебера обусловлена мутациями в генах мтДНК, кодирующих субъединицы
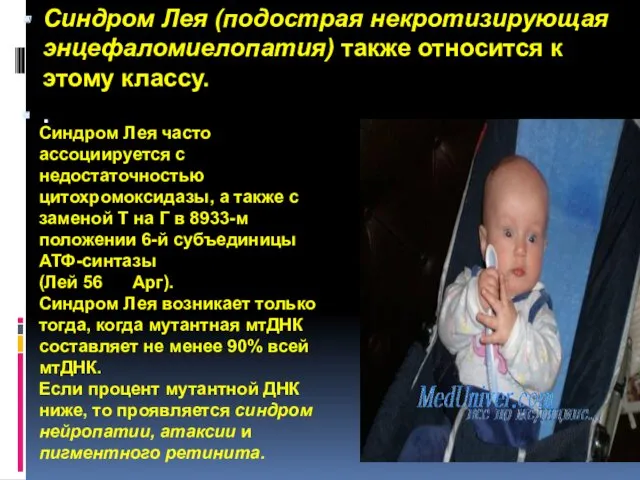
Синдром Лея (подострая некротизирующая энцефаломиелопатия) также относится к этому классу.
.
Синдром Лея (подострая некротизирующая энцефаломиелопатия) также относится к этому классу.
.
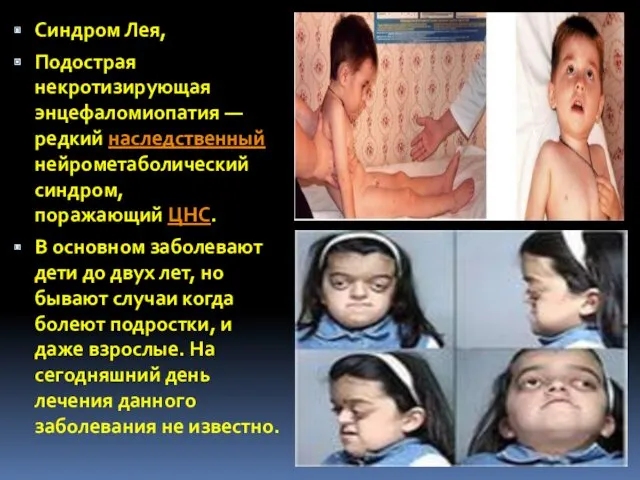
Синдром Лея,
Подострая некротизирующая энцефаломиопатия — редкий наследственный нейрометаболический синдром, поражающий ЦНС.
В основном заболевают
Синдром Лея,
Подострая некротизирующая энцефаломиопатия — редкий наследственный нейрометаболический синдром, поражающий ЦНС.
В основном заболевают

Синдром нейропатии, атаксии и пигментной дистрофии сетчатки (NARP) может проявляться как
Синдром нейропатии, атаксии и пигментной дистрофии сетчатки (NARP) может проявляться как
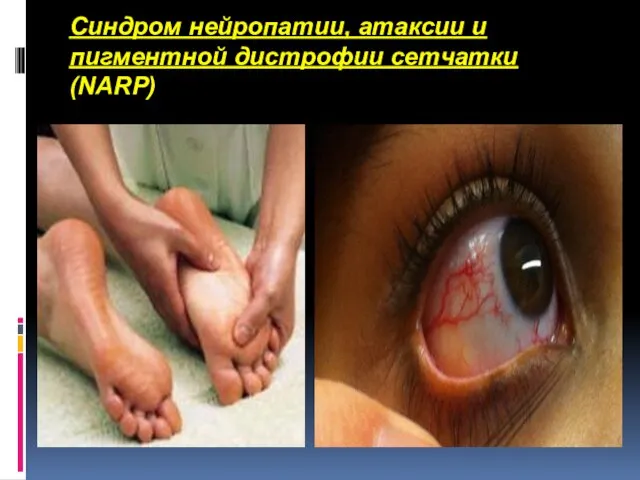
Синдром нейропатии, атаксии и пигментной дистрофии сетчатки (NARP)
Синдром нейропатии, атаксии и пигментной дистрофии сетчатки (NARP)

Синдром миоклонус-эпилепсии и рваных красных мышечных волокон (MERRF), который проявляется эпилепсией,
Синдром миоклонус-эпилепсии и рваных красных мышечных волокон (MERRF), который проявляется эпилепсией,
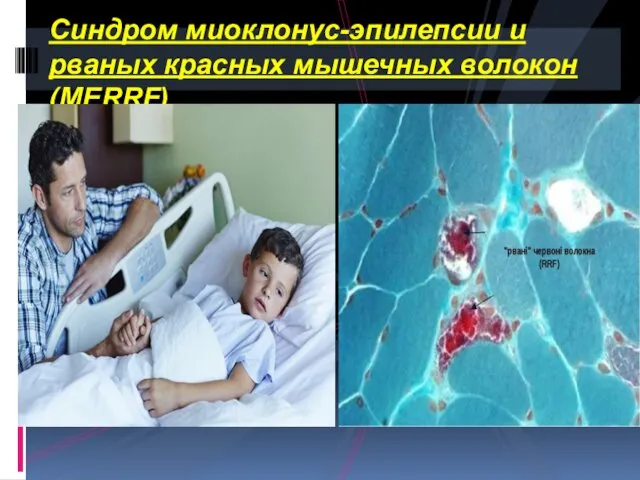
Синдром миоклонус-эпилепсии и рваных красных мышечных волокон (MERRF)
Синдром миоклонус-эпилепсии и рваных красных мышечных волокон (MERRF)

Синдром митохондриальной энцефаломиопатии и инсультоподобных эпизодов (MELAS)
Синдром митохондриальной энцефаломиопатии и инсультоподобных эпизодов (MELAS)

К МТБ, обусловленным делециями или дупликациями, относятся синдром Кернса-Сайра (миопатия, мозжечковые
К МТБ, обусловленным делециями или дупликациями, относятся синдром Кернса-Сайра (миопатия, мозжечковые

4.Болезни геномного импринтинга
геномный импринтинг и болезни импринтинга, это класс болезней,
4.Болезни геномного импринтинга
геномный импринтинг и болезни импринтинга, это класс болезней,
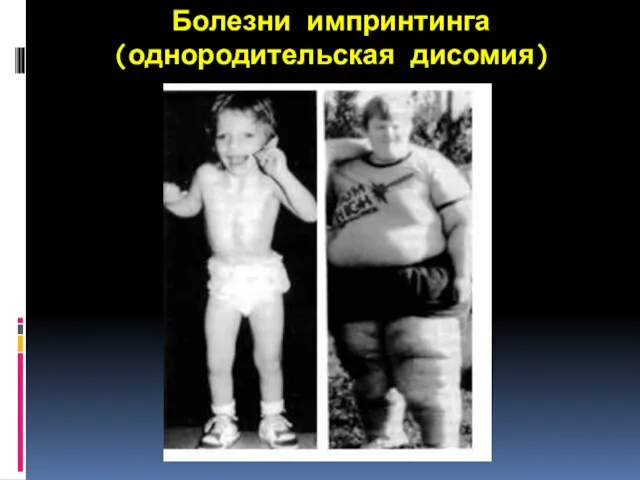
Болезни импринтинга (однородительская дисомия)
Болезни импринтинга (однородительская дисомия)

Синдром Х-ломкой хромосомы
Синдром Х-ломкой хромосомы

Геномный импринтинг – эпигенетическое явление, при котором наследуются изменения генной активности,
Геномный импринтинг – эпигенетическое явление, при котором наследуются изменения генной активности,

Первоначальный «отпечаток», созданный в половых клетках, служит основанием для дальнейших модификаций
Первоначальный «отпечаток», созданный в половых клетках, служит основанием для дальнейших модификаций

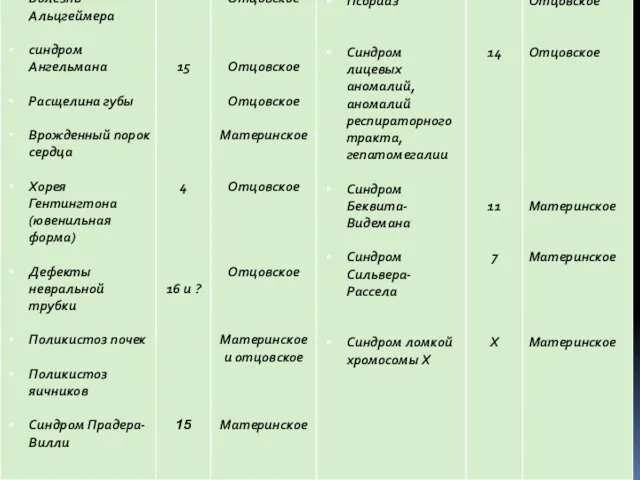
У человека эффект импринтинга обнаружен в связи с наличием в хромосомном
У человека эффект импринтинга обнаружен в связи с наличием в хромосомном
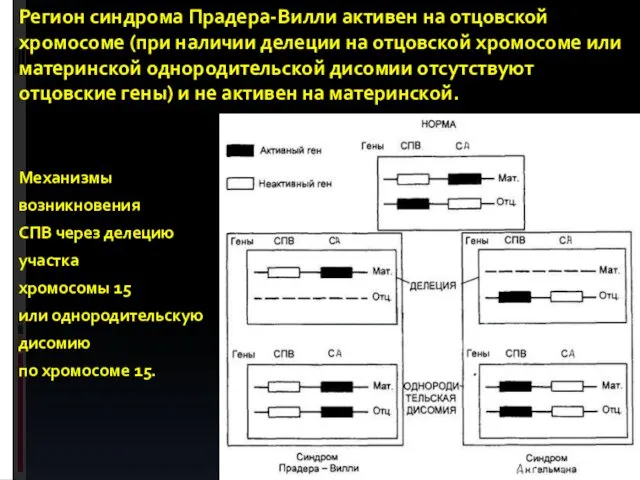
Регион синдрома Прадера-Вилли активен на отцовской хромосоме (при наличии делеции на
Регион синдрома Прадера-Вилли активен на отцовской хромосоме (при наличии делеции на

5. Мультифакторные болезни
мультифакторные болезни или болезни с наследственной предрасположенностью, так как
5. Мультифакторные болезни
мультифакторные болезни или болезни с наследственной предрасположенностью, так как

ПРИЧИНЫ БОЛЕЗНЕЙ С НАСЛЕДСТВЕННОЙ
ПРЕДРАСПОЛОЖЕННОСТЬЮ
ПРИЧИНЫ
СРЕДОВЫЕ
СЕМЕЙНЫЕ
ПОПУЛЯЦИОН-НЫЕ
ГЕНЕТИЧЕСКИЕ
ГЕНЫ ПРЕДРАСПО-ЛОЖЕННОСТИ
СТОХАСТИЧЕСКИЕ
(случайные)
ГЕНЕТИЧЕСКИЙ ФОН
ПРИЧИНЫ БОЛЕЗНЕЙ С НАСЛЕДСТВЕННОЙ
ПРЕДРАСПОЛОЖЕННОСТЬЮ
ПРИЧИНЫ
СРЕДОВЫЕ
СЕМЕЙНЫЕ
ПОПУЛЯЦИОН-НЫЕ
ГЕНЕТИЧЕСКИЕ
ГЕНЫ ПРЕДРАСПО-ЛОЖЕННОСТИ
СТОХАСТИЧЕСКИЕ
(случайные)
ГЕНЕТИЧЕСКИЙ ФОН

К ним относятся все хронические неинфекционные заболевания, такие как диабет, атеросклероз,
К ним относятся все хронические неинфекционные заболевания, такие как диабет, атеросклероз,

Одним из способов выявления генов, которые могут иметь отношение к предрасположенности
Одним из способов выявления генов, которые могут иметь отношение к предрасположенности

6. Заболевания, обусловленные преимущественно действием внешнесредовых факторов – это травмы, частые
6. Заболевания, обусловленные преимущественно действием внешнесредовых факторов – это травмы, частые
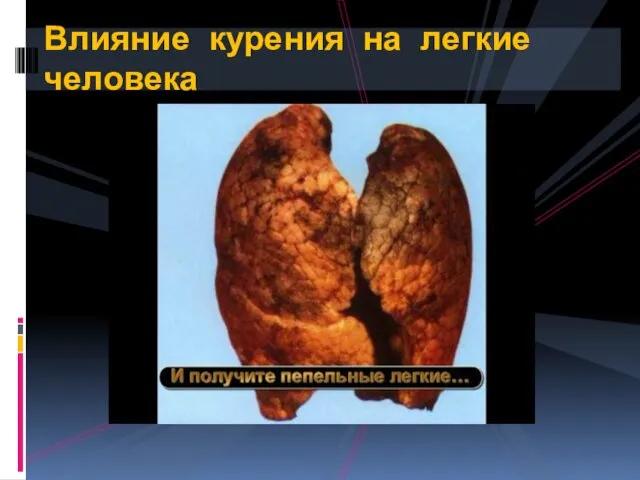
Влияние курения на легкие человека
Влияние курения на легкие человека
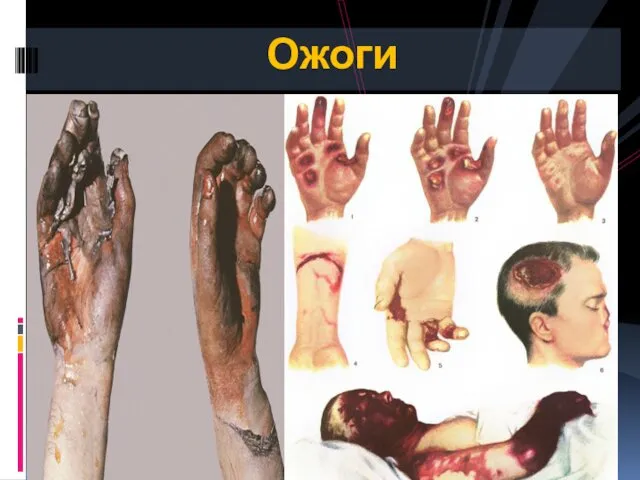
Ожоги
Ожоги
 Психология детей с сенсорными нарушениями
Психология детей с сенсорными нарушениями Лепра
Лепра Свойства материалов и их влияние на ткани зуба. Материаловедение. Лекция № 1. Тема 2
Свойства материалов и их влияние на ткани зуба. Материаловедение. Лекция № 1. Тема 2 Новости COVID-2019
Новости COVID-2019 Тамырлар жүйесі
Тамырлар жүйесі Острый живот у детей
Острый живот у детей Клещевой энцефалит
Клещевой энцефалит Генетикалық инженерия негіздері
Генетикалық инженерия негіздері Патология внешнего дыхания
Патология внешнего дыхания Босану физиологиясы. Босанудың басталу себептері
Босану физиологиясы. Босанудың басталу себептері Отбасын құруды жоспарлау
Отбасын құруды жоспарлау Кодекс этики в фармацевтической деятельности
Кодекс этики в фармацевтической деятельности Рентген-диагностика повреждений стопы
Рентген-диагностика повреждений стопы Волосатоклеточный лейкоз
Волосатоклеточный лейкоз Основы микрохирургии (микрососудистая)
Основы микрохирургии (микрососудистая) Токсоплазмоз ЦНС у пациента с ВИЧ-инфекцией. Разбор клинического случая
Токсоплазмоз ЦНС у пациента с ВИЧ-инфекцией. Разбор клинического случая Гипертоническая болезнь
Гипертоническая болезнь Кожные заболевания
Кожные заболевания Рак кожи
Рак кожи Хирургическое обследование. Общий осмотр
Хирургическое обследование. Общий осмотр Лекарственные средства, применяемые при АГ
Лекарственные средства, применяемые при АГ Этапы эндодонтического лечения
Этапы эндодонтического лечения Энцефалит - бас миының қабынуы
Энцефалит - бас миының қабынуы Здоровье ЖКТ от компании Natures Sunshine
Здоровье ЖКТ от компании Natures Sunshine Компоненты здорового образа жизни и пути их формирования
Компоненты здорового образа жизни и пути их формирования Электронды базалардан артериальды гипертензия және жүректің ишемиялық ауруы арасындағы байланыс жайлы мәліметтерді ғылыми
Электронды базалардан артериальды гипертензия және жүректің ишемиялық ауруы арасындағы байланыс жайлы мәліметтерді ғылыми Возбудители анаэробной инфекции
Возбудители анаэробной инфекции Эмбриогенез, анатомофизиологические особенности почек и мочевыделительной системы у детей
Эмбриогенез, анатомофизиологические особенности почек и мочевыделительной системы у детей