- Методы исследования врожденных пороков развития

Содержание
- 2. Классификация методов исследования Экспериментальный метод Клинические методы Морфологические методы Генетические методы
- 3. Особенности клинических методов Анамнез: наличие сходных пороков развития у родителей, сибсов или других родственников пробанда, близкородственные
- 4. Пример Изолированая расщелина губы и неба – полигенно наследуемый порок с риском повторения 3,5-4% Сочетание расщелины
- 5. день
- 6. Психическое развитие Олигофрения – частый признак множественных пороков развития Синдром Дауна
- 7. Анализ дерматоглифики – комплекса кожных узоров, расположенных на ладонях, подошвах и сгибательных поверхностях пальцев Анализируя признаки
- 8. Синдром Дауна
- 9. Генетические методы Генеалогический Цитогенетический Популяционно-статистический Близнецовый
- 10. Генеалогический метод – анализ родословных В ряде случаев помогает установить тип наследования порока. Родословная должна охватывать
- 11. Кариотип женщины
- 12. Доминантный тип наследования
- 13. Рецессивный тип наследования
- 14. Типы наследования Аутосомно-доминантный и аутосомно-рецессивный Х-сцепленный доминантный (пороки только у женщин) и Х-сцепленный рецессивный (пороки только
- 15. Аутосомно-доминантный тип наследования Хорея Хаттингтона Ахондроплазия Нейрофиброматоз Полидактилия Семейная ретинобластома
- 16. Поликистоз почек
- 17. Ахондроплазия
- 18. Нейрофиброматоз
- 19. Семейная ретинобластома
- 20. Аутосомно-рецессивный тип наследования Голубые глаза Серповидно-клеточная анемия Кистозный фиброз (муковисцидоз) БолезньТея-Сакса
- 21. Болезнь Тея-Сакса Болезнь проявляется в сильном нарушении моторных актов, восприятия и интеллектуальной деятельности. Для неё характерно
- 22. Серповидно-клеточная анемия
- 23. Х-сцепленный рецессивный тип наследования Гемофилия А/В Дальтонизм Синдром Гунтера (Хантера) Мышечная дистрофия Дюшена-Беккера
- 24. Синдром Хантера (Гунтера) – мукополисахаридоз II Дефект L-идуроносульфатсульфа- тазы, характеризующийся умеренно выраженной деформацией скелета, атрофией дисков
- 25. Миодистрофия Дюшена-Беккера Мышечная дистрофия Дюшена-Беккера (псевдогипертрофическая миопатия) - одно из самых частых нервно-мышечных заболеваний; связано с
- 26. Гемофилия А
- 27. Цитогенетический метод - определение полового хроматина или хромосомного набора ребенка (плода) с врожденными пороками или его
- 28. Морфологический метод Используется для исследования различных видов материала: патологоанатомического, эмбриологического, операционного, биопсийного
- 29. Дородовая диагностика ВПР Ультразвуковое исследование плода Определение кариотипа плода Амниоцентез с исследованием околоплодной жидкости и культивированием
- 30. Изменение наследственных структур (мутации) Генные мутации – изменение внутренней структуры отдельных генов. Хромосомные мутации – изменения
- 31. Критические периоды развития
- 32. Генные мутации Аутосомно-доминантный тип наследования: Ахондроплазия Синдром Марфана Нейрофиброматоз Хорея Хаттингтона и др.
- 33. Хорея Хаттингтона
- 34. Синдром Марфана
- 35. Ахондроплазия – локус 4р16
- 36. Ахондрогенез тип II –локус 5q31-34
- 37. Синдром Корнелии де Ланге – локус 3q26/17q23
- 38. Генные мутации Аутосомно-рецессивный тип наследования: Альбинизм Кистозный фиброз Фенилкетонурия Галактоземия
- 39. Альбинизм
- 40. Хромосомные мутации
- 41. Хромосомные мутации - синдром «кошачьего крика» (делеция 5р-) Частота – 1 на 45000 Специфический плач Умственная
- 42. Хромосомные мутации – кариотип синдрома «кошачьего крика
- 43. Prader-Willi синдром (делеция 15q11,2q12) Клиника: ожирение, гипотония, гипогонадизм Частота: 1 на 10,000 – 1 на 25,000
- 44. Частота - 1 на 7602 Пренатальная гипоплазия Тригоноцефалия, микроцефалия Гипотелоризм Аномалии скальпа Расщелина верхней губы и
- 45. Синдром Дауна – трисомия 21
- 46. Геномная мутация – синдром Дауна (трисомия 21) Частота – 1,25х10-3 Уплощение профиля лица Брахицефалия Монголоидный разрез
- 47. Синдром Эдвардса – трисомия 18
- 48. Анеуплоидия
- 49. Частота – 1 на 3000 Рудиментарные гонады Короткая складчатая шея Пороки сердца (25%) Аномалии почек Низкий
- 50. Частота – 1,13 на 1000 Евнухоидное телосложение Гинекомастия Микроорхидизм Оволосение по женскому типу Олигофрения в степени
- 51. Синдром Якобса - ХУУ
- 53. Скачать презентацию

Классификация методов исследования
Экспериментальный метод
Клинические методы
Морфологические методы
Генетические методы
Классификация методов исследования
Экспериментальный метод
Клинические методы
Морфологические методы
Генетические методы

Особенности клинических методов
Анамнез: наличие сходных пороков развития у родителей, сибсов или
Особенности клинических методов
Анамнез: наличие сходных пороков развития у родителей, сибсов или

Пример
Изолированая расщелина губы и неба – полигенно наследуемый порок с риском
Пример
Изолированая расщелина губы и неба – полигенно наследуемый порок с риском
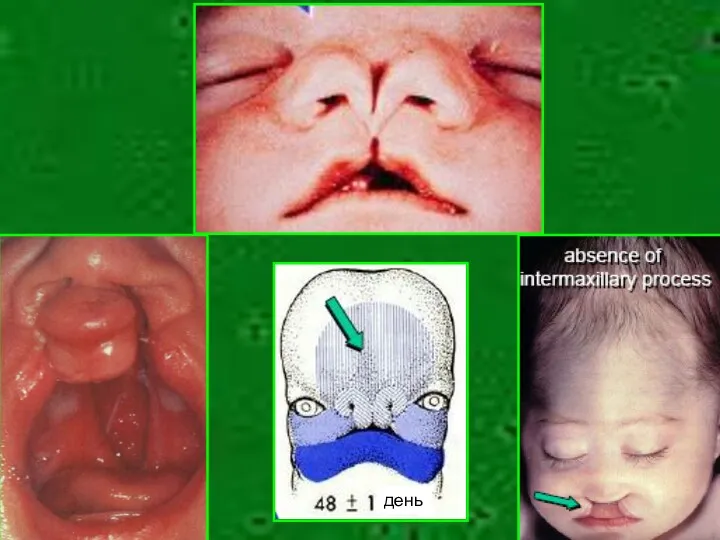
день
день

Психическое развитие
Олигофрения – частый признак множественных пороков развития
Синдром Дауна
Психическое развитие
Олигофрения – частый признак множественных пороков развития
Синдром Дауна
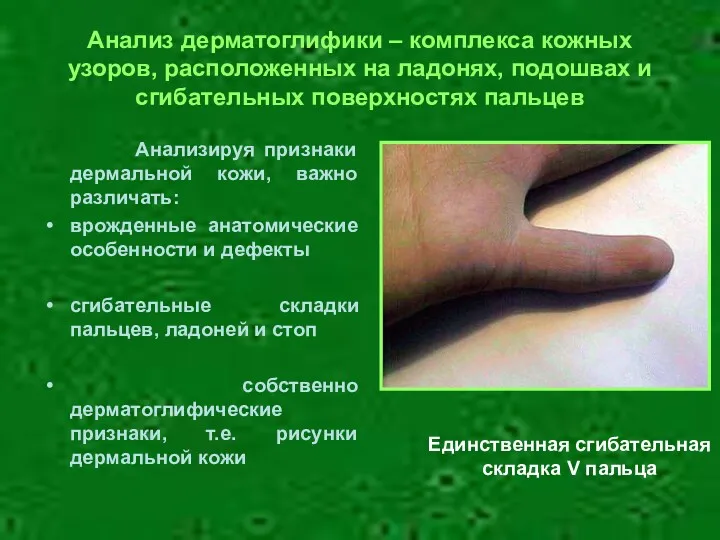
Анализ дерматоглифики – комплекса кожных узоров, расположенных на ладонях, подошвах и
Анализ дерматоглифики – комплекса кожных узоров, расположенных на ладонях, подошвах и
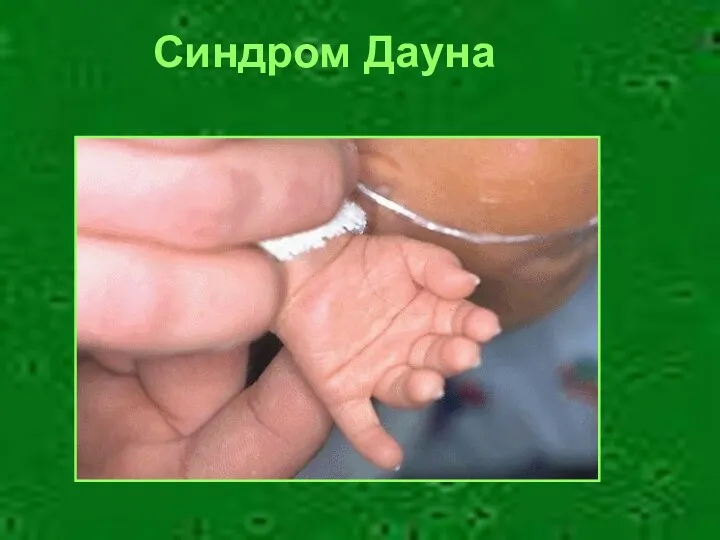
Синдром Дауна
Синдром Дауна

Генетические методы
Генеалогический
Цитогенетический
Популяционно-статистический
Близнецовый
Генетические методы
Генеалогический
Цитогенетический
Популяционно-статистический
Близнецовый

Генеалогический метод – анализ родословных
В ряде случаев помогает установить тип наследования
Генеалогический метод – анализ родословных
В ряде случаев помогает установить тип наследования

Кариотип женщины
Кариотип женщины
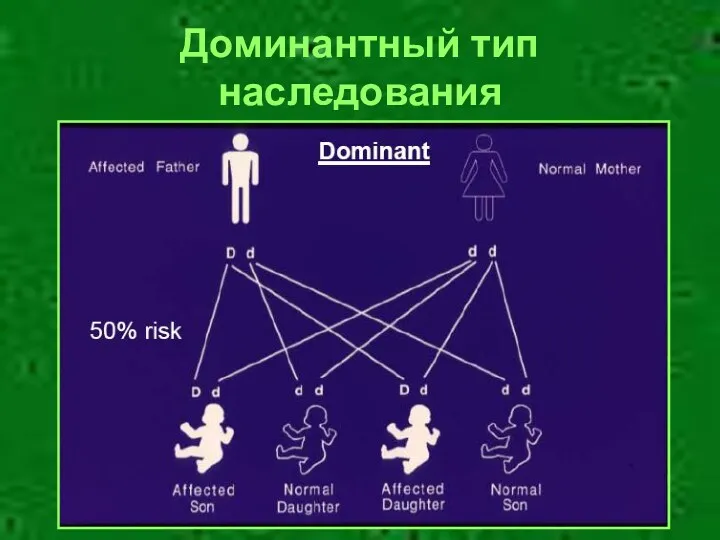
Доминантный тип наследования
Доминантный тип наследования
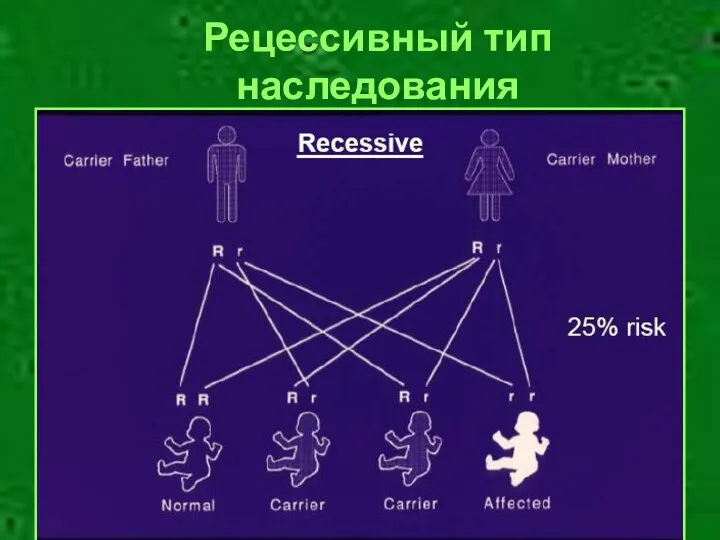
Рецессивный тип наследования
Рецессивный тип наследования
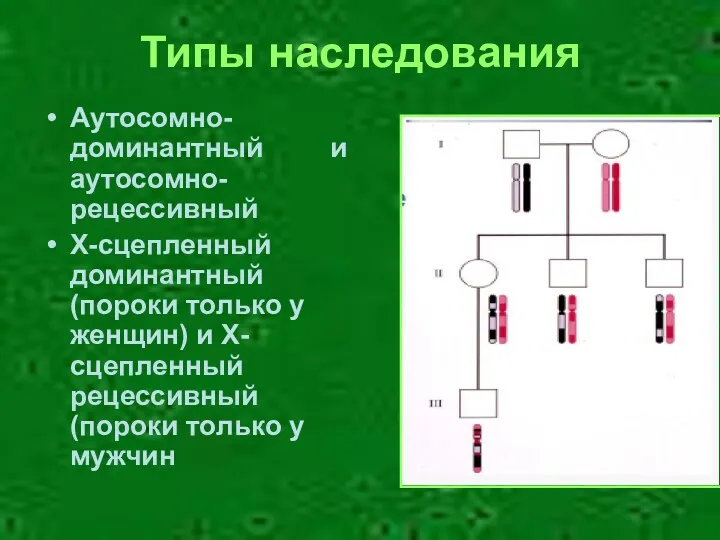
Типы наследования
Аутосомно-доминантный и аутосомно-рецессивный
Х-сцепленный доминантный (пороки только у женщин) и Х-сцепленный
Типы наследования
Аутосомно-доминантный и аутосомно-рецессивный
Х-сцепленный доминантный (пороки только у женщин) и Х-сцепленный
Аутосомно-доминантный тип наследования
Хорея Хаттингтона
Ахондроплазия
Нейрофиброматоз
Полидактилия
Семейная
ретинобластома
Аутосомно-доминантный тип наследования
Хорея Хаттингтона
Ахондроплазия
Нейрофиброматоз
Полидактилия
Семейная
ретинобластома
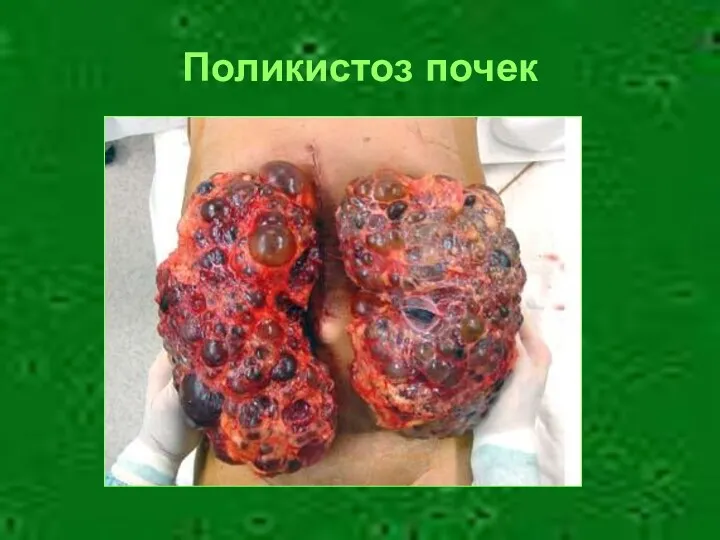
Поликистоз почек
Поликистоз почек

Ахондроплазия
Ахондроплазия
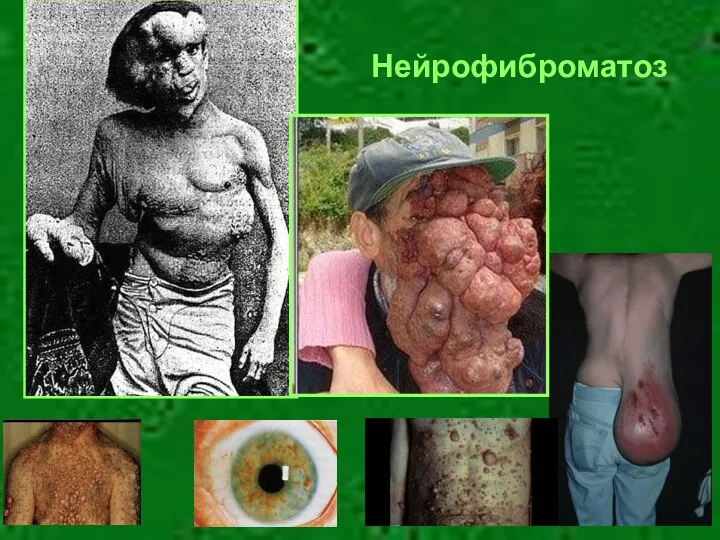
Нейрофиброматоз
Нейрофиброматоз
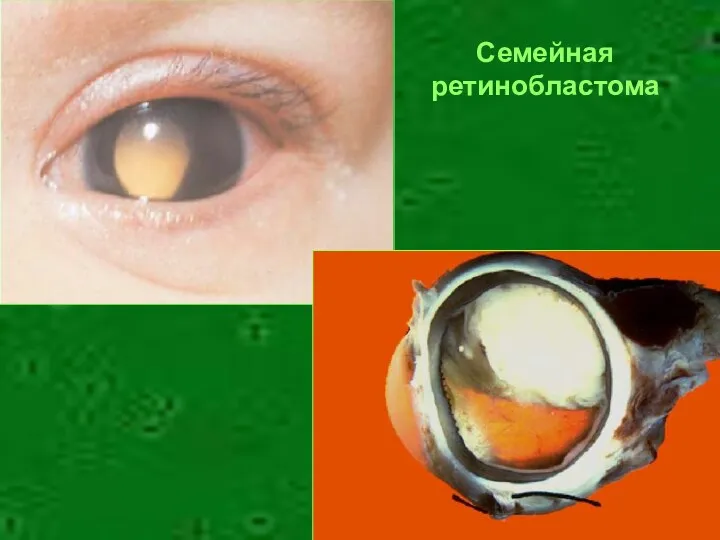
Семейная
ретинобластома
Семейная
ретинобластома

Аутосомно-рецессивный тип наследования
Голубые глаза
Серповидно-клеточная анемия
Кистозный фиброз (муковисцидоз)
БолезньТея-Сакса
Аутосомно-рецессивный тип наследования
Голубые глаза
Серповидно-клеточная анемия
Кистозный фиброз (муковисцидоз)
БолезньТея-Сакса
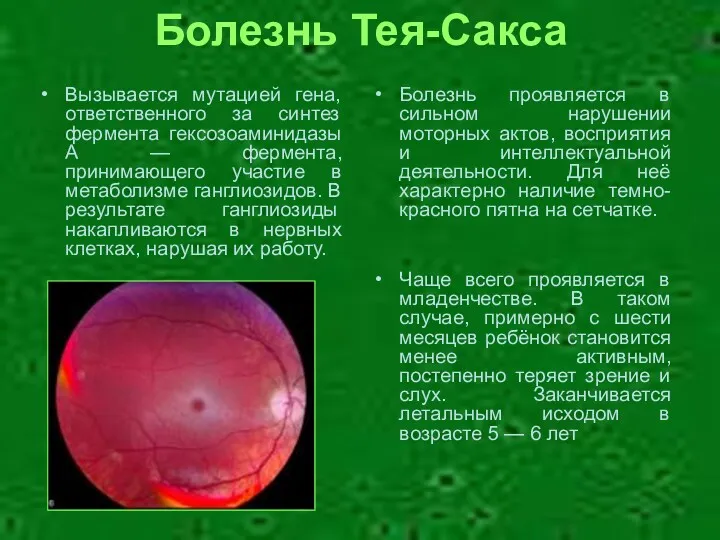
Болезнь Тея-Сакса
Болезнь проявляется в сильном нарушении моторных актов, восприятия и интеллектуальной
Болезнь Тея-Сакса
Болезнь проявляется в сильном нарушении моторных актов, восприятия и интеллектуальной
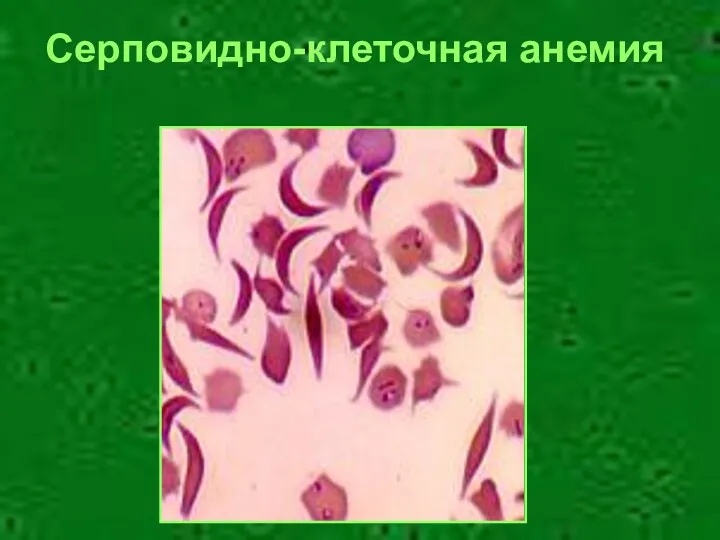
Серповидно-клеточная анемия
Серповидно-клеточная анемия

Х-сцепленный рецессивный тип наследования
Гемофилия А/В
Дальтонизм
Синдром Гунтера (Хантера)
Мышечная дистрофия Дюшена-Беккера
Х-сцепленный рецессивный тип наследования
Гемофилия А/В
Дальтонизм
Синдром Гунтера (Хантера)
Мышечная дистрофия Дюшена-Беккера

Синдром Хантера (Гунтера) – мукополисахаридоз II
Дефект L-идуроносульфатсульфа-
тазы, характеризующийся умеренно выраженной
Синдром Хантера (Гунтера) – мукополисахаридоз II
Дефект L-идуроносульфатсульфа-
тазы, характеризующийся умеренно выраженной

Миодистрофия Дюшена-Беккера
Мышечная дистрофия Дюшена-Беккера (псевдогипертрофическая миопатия) - одно из самых частых
Миодистрофия Дюшена-Беккера
Мышечная дистрофия Дюшена-Беккера (псевдогипертрофическая миопатия) - одно из самых частых
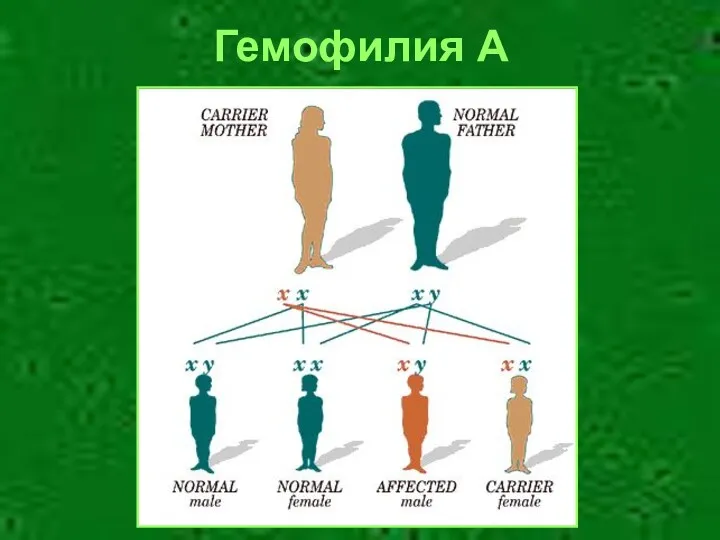
Гемофилия А
Гемофилия А

Цитогенетический метод - определение полового хроматина или хромосомного набора ребенка (плода)
Цитогенетический метод - определение полового хроматина или хромосомного набора ребенка (плода)

Морфологический метод
Используется для исследования различных видов материала: патологоанатомического, эмбриологического, операционного, биопсийного
Морфологический метод
Используется для исследования различных видов материала: патологоанатомического, эмбриологического, операционного, биопсийного

Дородовая диагностика ВПР
Ультразвуковое исследование плода
Определение кариотипа плода
Амниоцентез с исследованием околоплодной
Дородовая диагностика ВПР
Ультразвуковое исследование плода
Определение кариотипа плода
Амниоцентез с исследованием околоплодной

Изменение наследственных структур (мутации)
Генные мутации – изменение внутренней структуры отдельных генов.
Хромосомные
Изменение наследственных структур (мутации)
Генные мутации – изменение внутренней структуры отдельных генов.
Хромосомные
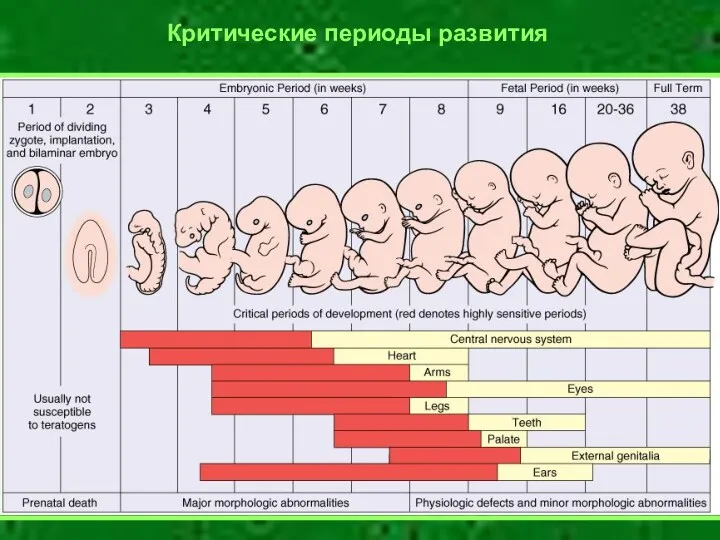
Критические периоды развития
Критические периоды развития

Генные мутации
Аутосомно-доминантный тип наследования:
Ахондроплазия
Синдром Марфана
Нейрофиброматоз
Хорея Хаттингтона и др.
Генные мутации
Аутосомно-доминантный тип наследования:
Ахондроплазия
Синдром Марфана
Нейрофиброматоз
Хорея Хаттингтона и др.
Хорея Хаттингтона
Хорея Хаттингтона

Синдром Марфана
Синдром Марфана
Ахондроплазия – локус 4р16
Ахондроплазия – локус 4р16
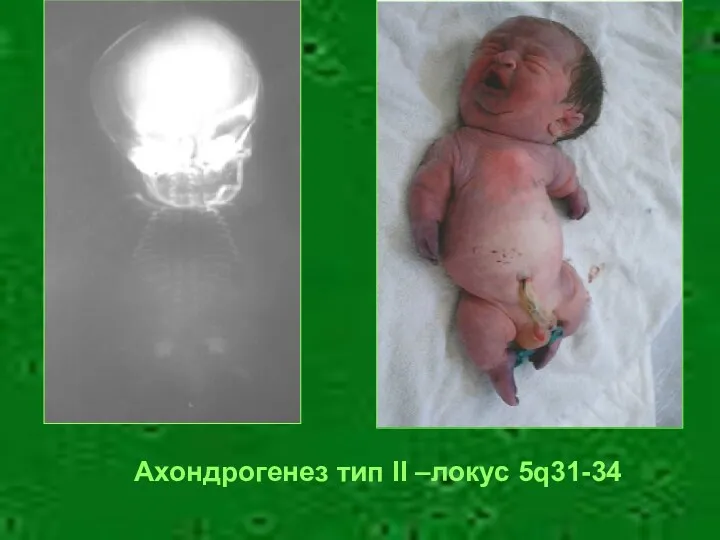
Ахондрогенез тип II –локус 5q31-34
Ахондрогенез тип II –локус 5q31-34

Синдром Корнелии де Ланге – локус 3q26/17q23
Синдром Корнелии де Ланге – локус 3q26/17q23

Генные мутации
Аутосомно-рецессивный тип наследования:
Альбинизм
Кистозный фиброз
Фенилкетонурия
Галактоземия
Генные мутации
Аутосомно-рецессивный тип наследования:
Альбинизм
Кистозный фиброз
Фенилкетонурия
Галактоземия
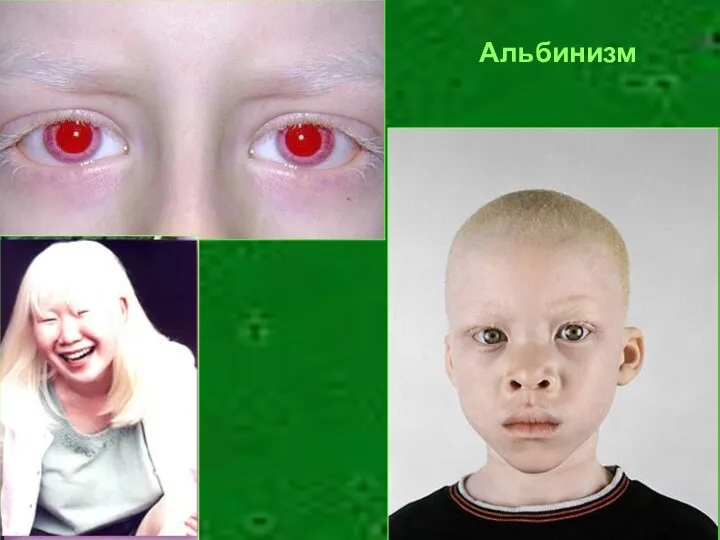
Альбинизм
Альбинизм

Хромосомные мутации
Хромосомные мутации

Хромосомные мутации -
синдром «кошачьего крика» (делеция 5р-)
Частота – 1
Хромосомные мутации -
синдром «кошачьего крика» (делеция 5р-)
Частота – 1

Хромосомные мутации –
кариотип синдрома «кошачьего крика
Хромосомные мутации –
кариотип синдрома «кошачьего крика

Prader-Willi синдром (делеция 15q11,2q12)
Клиника: ожирение, гипотония, гипогонадизм
Частота: 1 на 10,000 –
Prader-Willi синдром (делеция 15q11,2q12)
Клиника: ожирение, гипотония, гипогонадизм
Частота: 1 на 10,000 –
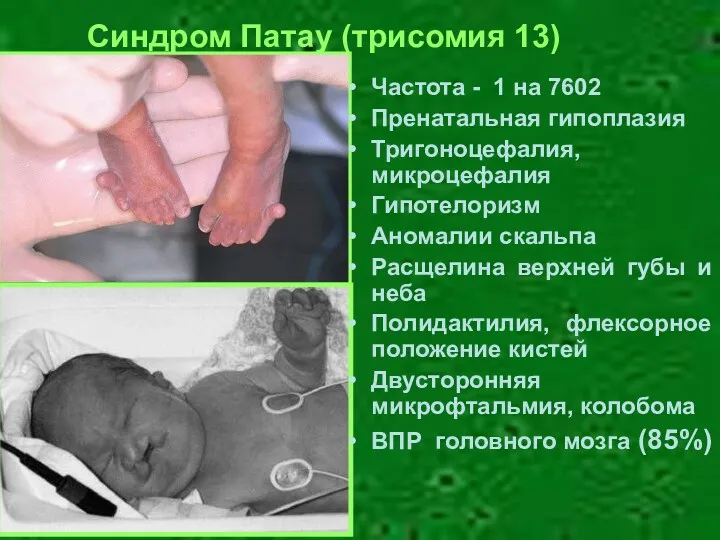
Частота - 1 на 7602
Пренатальная гипоплазия
Тригоноцефалия, микроцефалия
Гипотелоризм
Аномалии скальпа
Расщелина верхней губы и
Частота - 1 на 7602
Пренатальная гипоплазия
Тригоноцефалия, микроцефалия
Гипотелоризм
Аномалии скальпа
Расщелина верхней губы и
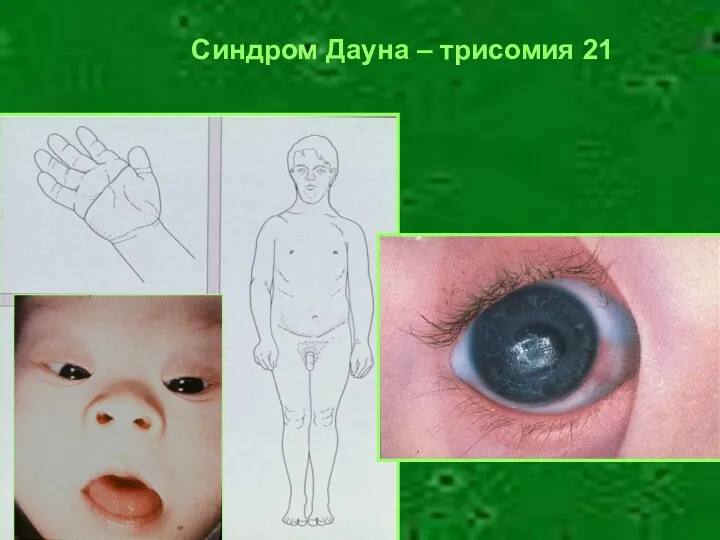
Синдром Дауна – трисомия 21

Геномная мутация – синдром Дауна (трисомия 21)
Частота – 1,25х10-3
Уплощение профиля лица
Брахицефалия
Монголоидный
Геномная мутация – синдром Дауна (трисомия 21)
Частота – 1,25х10-3
Уплощение профиля лица
Брахицефалия
Монголоидный
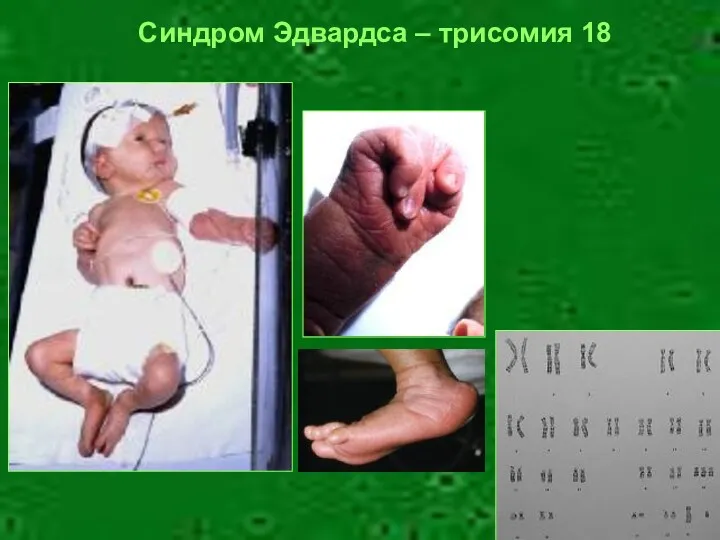
Синдром Эдвардса – трисомия 18
Синдром Эдвардса – трисомия 18
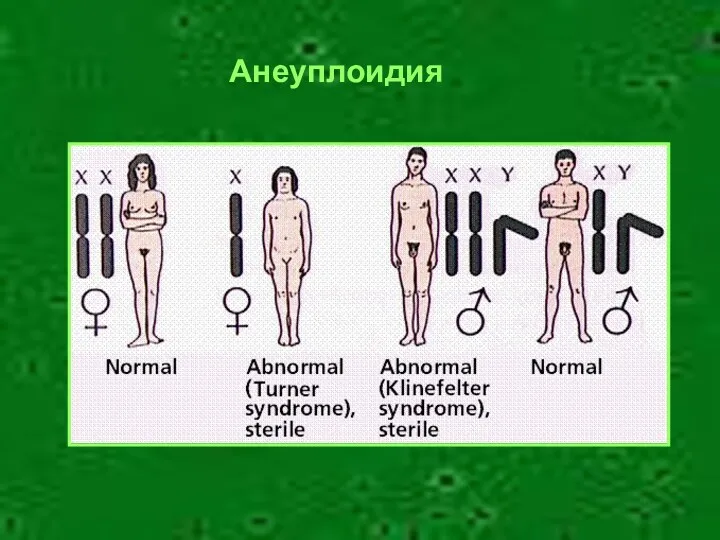
Анеуплоидия
Анеуплоидия
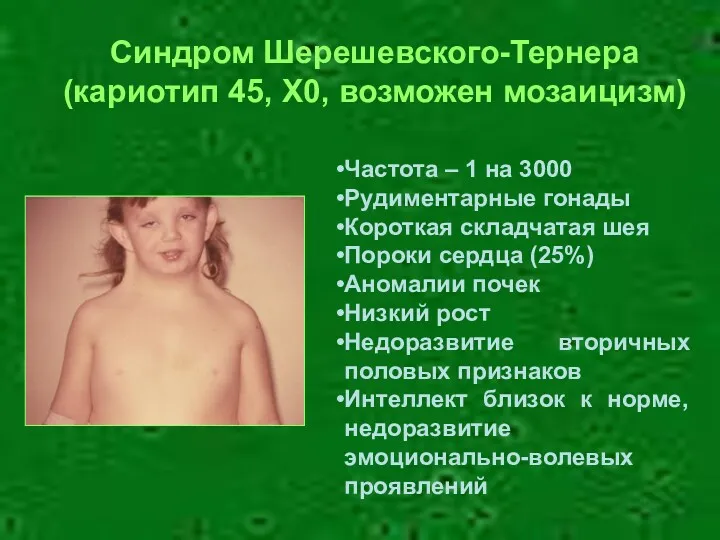
Частота – 1 на 3000
Рудиментарные гонады
Короткая складчатая шея
Пороки сердца (25%)
Аномалии почек
Низкий
Частота – 1 на 3000
Рудиментарные гонады
Короткая складчатая шея
Пороки сердца (25%)
Аномалии почек
Низкий
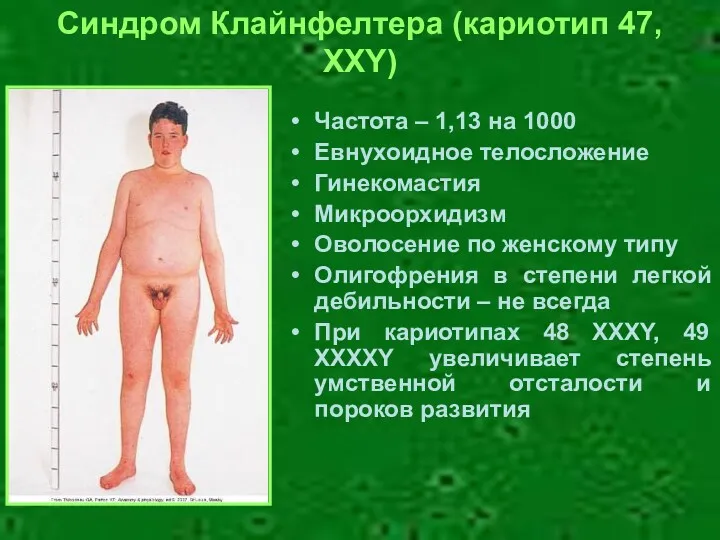
Частота – 1,13 на 1000
Евнухоидное телосложение
Гинекомастия
Микроорхидизм
Оволосение по женскому типу
Олигофрения в степени
Частота – 1,13 на 1000
Евнухоидное телосложение
Гинекомастия
Микроорхидизм
Оволосение по женскому типу
Олигофрения в степени

Синдром Якобса - ХУУ
Синдром Якобса - ХУУ
 Балалар мен жасөспірімдердің дене тәрбиесі мен шынығуының физикалық-гигиеналық негіздері
Балалар мен жасөспірімдердің дене тәрбиесі мен шынығуының физикалық-гигиеналық негіздері Этика и эстетика в ортопедической стоматологии
Этика и эстетика в ортопедической стоматологии Комитетінің құрылымы және мемлекеттік санитарлық эпидемиологиялық жүйесінің жұмысының қазіргі кездегі бағыттары
Комитетінің құрылымы және мемлекеттік санитарлық эпидемиологиялық жүйесінің жұмысының қазіргі кездегі бағыттары Аптечка первой медицинской помощи
Аптечка первой медицинской помощи Отравления алкоголем, наркотическими средствами, лекарственными препаратами. Укусы змей. Аллергические реакции
Отравления алкоголем, наркотическими средствами, лекарственными препаратами. Укусы змей. Аллергические реакции Введение в фармакологию. Лекция №1
Введение в фармакологию. Лекция №1 Токсокароз плотоядных. Механизмы и побочные действия антигельминтиков
Токсокароз плотоядных. Механизмы и побочные действия антигельминтиков Гастроэзофагальная рефлюксная болезнь (ГЭРБ)
Гастроэзофагальная рефлюксная болезнь (ГЭРБ) Эпидемиология. Туберкулез және АИВ
Эпидемиология. Туберкулез және АИВ Клинический разбор
Клинический разбор Паразитические инфузории и споровики
Паразитические инфузории и споровики Организация наблюдения за новорожденными на педиатрическом участке
Организация наблюдения за новорожденными на педиатрическом участке Гипогонадизм. Гирсутизм
Гипогонадизм. Гирсутизм Зубы. Твердые и мягкие ткани зуба. Поддерживающий аппарат зуба
Зубы. Твердые и мягкие ткани зуба. Поддерживающий аппарат зуба Туберкулинодиагностика
Туберкулинодиагностика Клиническая физиология водно-солевого обмена
Клиническая физиология водно-солевого обмена Возбудители особо опасных и зоонозных инфекций
Возбудители особо опасных и зоонозных инфекций Инфаркт миокарда
Инфаркт миокарда Протезирование дефектов зубных рядов пластмассовыми комбинированными и цельнокерамическими коронками
Протезирование дефектов зубных рядов пластмассовыми комбинированными и цельнокерамическими коронками The heart sounds
The heart sounds Медицинская статистика
Медицинская статистика Классификация шизофрении
Классификация шизофрении Повреждение мягких тканей у детей (ушибы, раны). Принципы хирургической обработки
Повреждение мягких тканей у детей (ушибы, раны). Принципы хирургической обработки Моногенные болезни
Моногенные болезни Укрепление иммунитета
Укрепление иммунитета Организация сестринского ухода за детьми с дистрофиями
Организация сестринского ухода за детьми с дистрофиями Нервная система человека
Нервная система человека Обращение с медицинскими отходами
Обращение с медицинскими отходами