- Нервно-мышечные заболевания

Содержание
- 2. ОПРЕДЕЛЕНИЕ Нервно-мышечные заболевания – болезни с поражением нейронов, их аксонов, синапсов или самих мышц Болезнь мышечной
- 3. ПРИЗНАКИ ПОРАЖЕНИЯ Гипотония Гипорефлексия Гипотрофия
- 4. КРУГ НЕРВНО-МЫШЕЧНЫХ ЗАБОЛЕВАНИЙ Наследственные заболевания Инфекционные Воспалительные Паранеопластические При соматических заболеваниях При эндокринных заболеваниях и др.
- 5. НЕРВНО-МЫШЕЧНАЯ ЕДИНИЦА А Б В Г
- 6. КЛАССИФИКАЦИЯ А Заболевания, связанные с поражением передних рогов спинного мозга Б Болезни, связанные с поражением периферических
- 7. ПОРАЖЕНИЕ ПЕРЕДНЕГО РОГА Только двигательные нарушения + фасцикуляции Отсутствуют чувствительные нарушения
- 8. ПОРАЖЕНИЕ ПЕРИФЕРИЧЕСКИХ НЕРВОВ Дистальные парезы (кисти, стопы) В подавляющем большинстве случаев + расстройства чувствительности + вегетативные
- 9. БОЛЕЗНИ СИНАПСА Патологическая мышечная утомляемость
- 10. МЫШЕЧНОЕ ПОРАЖЕНИЕ Преимущественное поражение проксимальных отделов конечностей (тазовый, плечевой пояс) Отсутствуют чувствительные нарушения
- 11. ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ ЭМГ Глобальная (поверхностные электроды, суммарная активность) Игольчатая (активность отдельного мышечного волокна) Скорость проведения возбуждения
- 12. ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ Исследование ферментов крови ↑ КФК - креатинфосфокиназа ↑ ЛДГ – лактатдегидрогеназа Исследование электролитов крови
- 13. ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ Биопсия мышц (гистохимическое исследование) Составление родословных таблиц
- 14. БОЛЕЗНИ ПЕРЕДНЕГО РОГА Инфекционные Клещевой энцефалит Полиомиелит (болезнь Гейне-Медина)- эпидемический детский паралич вирусной этиологии Полиомиелитоподобные заболевания
- 15. БОЛЕЗНИ ПЕРЕДНЕГО РОГА Дегенеративные заболевания Боковой амиотрофический склероз (БАС) – болезнь Шарко дегенерация боковых столбов поражение
- 16. БОКОВОЙ АМИОТРОФИЧЕСКИЙ СКЛЕРОЗ Пирамидный путь Передний рог
- 17. УРОВНИ ПОРАЖЕНИЯ ПРИ БАС Шейное утолщение Поясничное утолщение Бульбарный отдел ствола головного мозга
- 18. КЛИНИКА БАС Смешанные парезы в руках и/или ногах: признаки вовлечения переднего рога (фасцикуляции, атрофии) признаки поражения
- 19. ТЕЧЕНИЕ БАС Течение хроническое или подострое Варианты течения: как правило, восходящее, но может быть начало с
- 20. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ - СПИНАЛЬНЫЕ АМИОТРОФИИ Ранняя детская форма Вернига-Гофманна Юношеская форма Кугельберга-Веландер Бульбо-спинальная амиотрофия Кеннеди
- 21. РАННЯЯ ДЕТСКАЯ ФОРМА ВЕРНИГА - ГОФМАННА Описана в 1891 году Острая злокачественная инфантильная спинальная амиотрофия Тип
- 22. КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА Период внутриутробного развития – отсутствие или слабое шевеление плода у 1/3 матерей
- 23. КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА «вялый ребенок»
- 24. КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА Мышечная гипотония
- 25. ЮНОШЕСКАЯ ФОРМА КУГЕЛЬБЕРГА - ВЕЛАНДЕР Описана в 1956 году Течение доброкачественное Тип наследования – аутосомно-рецессивный
- 26. КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ КУГЕЛЬБЕРГА-ВЕЛАНДЕР Начало заболевания в 2 -15 лет (в среднем в 5 лет) Очень
- 27. СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ Тяжелая атрофия мышц плечевого пояса
- 28. БУЛЬБО-СПИНАЛЬНАЯ АМИОТРОФИЯ КЕННЕДИ Описана в 1968 году Доброкачественное течение Тип наследования – рецессивный, сцепленный с Х-хромосомой
- 29. СЦЕПЛЕННОЕ С Х-ХРОМОСОМОЙ РЕЦЕССИВНОЕ НАСЛЕДОВАНИЕ Мать-носитель Здоровый отец Здоровый мужчина Здоровая женщина Больной мужчина Женщина-носитель Хх
- 30. КЛИНИКА БУЛЬБО-СПИНАЛЬНОЙ АМИОТРОФИИ КЕННЕДИ Начало в зрелом возрасте (4-я декада) Проксимальная слабость в руках, ногах Бульбарный
- 31. БОЛЕЗНИ, СВЯЗАННЫЕ С ПОРАЖЕНИЕМ ПЕРИФЕРИЧЕСКИХ НЕРВОВ Общие признаки: периферические парезы полиневритический тип нарушения чувствительности вегетативные нарушения
- 32. ВИДЫ ПОЛИНЕЙРОПАТИЙ Аксональные полинейропатии: при дефиците тиамина, рибофлавина при отравлении мышьяком лекарственные полинейропатии (нитрофуран, изониазид, пиридоксин
- 33. НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ Группа наследственных сенсомоторных полинейропатий Описана Ж.Шарко, П.Мари в 1886 г. и Г.Тус в
- 34. КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ Начало с ног: постепенно нарастает слабость и атрофии мышц голеней, мелких мышц
- 35. КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ А Б А – дистальные атрофии Б – полиневритический тип нарушения чувствительности
- 36. Атрофия мышц кисти, «когтистая лапа» Невральная амиотрофия Шарко-Мари
- 37. Стопа Фридрайха, «полая» стопа Невральная амиотрофия Шарко-Мари
- 38. НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ Типы наследования: Аутосомно-доминантный Аутосомно-рецессивный Сцепленный с Х-хромосомой
- 39. ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА Синдром патологической мышечной утомляемости – нарастание пареза (слабости) к вечеру
- 40. ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА Ботулизм Миастения Миастенические синдромы
- 41. ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА Ботулизм возникает преходящая блокада пресинаптических структур, нарушается холинергическая трансмиссия связан с воздействием токсина
- 42. ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА Миастения (Myasthenia gravis) Заболевание аутоиммунной природы Вырабатываются антитела к белку ацетилхолиновых рецепторов (есть
- 43. КЛИНИКА МИАСТЕНИИ Начало, как правило, с глазных мышц (птоз, двоение, сонное выражение лица) Часто начало с
- 44. КЛИНИЧЕСКИЕ ПРОБЫ, ВЫЯВЛЯЮЩИЕ ПАТОЛОГИЧЕСКУЮ МЫШЕЧНУЮ УТОМЛЯЕМОСТЬ Фиксировать взор вверх – 30 секунд (больной устает) Громко считать
- 45. ТЕЧЕНИЕ МИАСТЕНИИ Характерно течение с ремиссиями В 20% наблюдаются миастенические кризы (генерализованная мышечная слабость, бульбарные, дыхательные
- 46. ЛЕЧЕНИЕ МИАСТЕНИИ Антихолинэстеразные прозерин – быстрого действия калимин – медленного Преднизолон (больным старше 50 лет) Плазмаферез
- 47. МИАСТЕНИЧЕСКИЕ СИНДРОМЫ Синдром Ламберта-Итона – паранеопластический синдром Наблюдается у мужчин старше 40 лет при бронхогенном раке
- 48. МИАСТЕНИЧЕСКИЕ СИНДРОМЫ Клиника не страдают глазные мышцы нет реакции на антихолинэстеразные препараты миастенический синдром может опережать
- 49. МИАСТЕНИЧЕСКИЕ СИНДРОМЫ Другие причины Заболевания щитовидной железы (аутоиммунные) Интоксикация лекарственными препаратами: неомицин гентамицин Д-пенициламин (при лечении
- 50. БОЛЕЗНИ, СВЯЗАННЫЕ С ПОРАЖЕНИЕМ МЫШЦ (МИОПАТИИ) Общие признаки: Отсутствие чувствительных и вегетативных нарушений Поражение проксимальных отделов
- 51. ПОЛИМИОЗИТ Не наследственное (аутоиммунное) заболевание Нарушение клеточного и гуморального иммунитета («воспалительная миопатия») Может протекать с кожными
- 52. КЛИНИКА ПОЛИМИОЗИТА Начало – острое или подострое Мышечная слабость Недомогание, артралгии, миалгии Повышение температуры тела Поражение
- 53. ПОЛИМИОЗИТ Данные дополнительного исследования: Уровень КФК не коррелирует с тяжестью ↑ миоглобин в сыворотке крови ↑СОЭ
- 54. ВОСПАЛИТЕЛЬНАЯ МИОПАТИЯ (ПОЛИМИОЗИТ) I Биопсия мышцы Воспалительные изменения в мышце
- 55. НАСЛЕДСТВЕННЫЕ МИОДИСТРОФИИ (МИОПАТИИ) Известно много форм и вариантов Псевдогипертрофическая миодистрофия Дюшена Миодистрофия Ландузи-Дежерина Миодистрофия Эрба
- 56. Неравномерность диаметра мышечных волокон, разрастание соединительной и жировой тканей ПЕРВИЧНЫЕ АМИОТРОФИИ (МИОПАТИИ) гистологическая картина четырехглавой мышцы
- 57. ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МИОДИСТРОФИЯ ДЮШЕНА Описана в 1861 году Дюшеном В 1879 году Говерс обобщил материал: 21 больной
- 58. КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА Первые признаки появляются с момента начала ходьбы (близко колени, ноги ставятся на
- 59. КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА Умственная отсталость – 30%. ЭМГ – признаки первично-мышечного поражения. Биопсия мышц –
- 60. КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА псевдогипертрофии
- 61. Мальчик 5 лет Наблюдаются псевдогипертрофии мышц, лордоз ПСЕВДОГИПЕРТРОФИЧЕСКАЯ ФОРМА ДЮШЕНА
- 62. СИМПТОМ «ВСТАВАНИЯ ЛЕСЕНКОЙ» ПРИ МИОПАТИИ
- 63. МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА Описана в 1884 -1886 г.г. (Дежерин, Ландузи) Частота встречаемости–0,4 на 100 000 населения Тип
- 64. КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА Дебют в 15-25 лет (≈20 лет) Слабость и атрофии мышц лица, плечевого пояса,
- 65. КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА Слабость мышц лица: глазные щели не смыкаются ночью больные не могут свистеть, пить
- 66. КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА Атрофия и слабость мышц плечевого и тазового поясов: крыловидные лопатки, гипотрофия передней лестничной
- 67. КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА Интеллект не страдает КФК↑ у 50 - 80% ЛДГ↑ у 20% Альдолаза↑ у
- 68. ПЛЕЧЕЛОПАТОЧНО-ЛИЦЕВАЯ МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА Поражение мышц лица и плечевого пояса Мальчик 13 лет, болен с 7 лет,
- 69. ЛОПАТОЧНО-ПЕРОНЕАЛЬНАЯ ФОРМА Выражена атрофия мышц плечевого пояса и перонеальных мышц Мальчик 15 лет, болен с 7
- 70. МИОДИСТРОФИЯ ЭРБА-РОТА Описана в 1884 году Эрбом Тип наследования аутосомно-рецессивный Экспрессивность гена у членов семьи разная
- 71. КЛИНИКА МИОДИСТРОФИИ ЭРБА Дебют во 2-м десятилетии, но может быть и в детстве и после 30
- 72. КЛИНИКА МИОДИСТРОФИИ ЭРБА Форма Лейдена-Мебиуса начало с проксимальных отделов ног Форма Эрба начало с плечевого пояса+спина,
- 73. ТАЗОВО-БЕДРЕННАЯ ФОРМА МИОПАТИИ ЭРБА Больной 17 лет, болен с 6 лет Атрофия мышц тазового и плечевого
- 75. Скачать презентацию

ОПРЕДЕЛЕНИЕ
Нервно-мышечные заболевания – болезни с поражением нейронов, их аксонов, синапсов или
ОПРЕДЕЛЕНИЕ
Нервно-мышечные заболевания – болезни с поражением нейронов, их аксонов, синапсов или

ПРИЗНАКИ ПОРАЖЕНИЯ
Гипотония
Гипорефлексия
Гипотрофия
ПРИЗНАКИ ПОРАЖЕНИЯ
Гипотония
Гипорефлексия
Гипотрофия

КРУГ НЕРВНО-МЫШЕЧНЫХ ЗАБОЛЕВАНИЙ
Наследственные заболевания
Инфекционные
Воспалительные
Паранеопластические
При соматических заболеваниях
При эндокринных заболеваниях и др.
КРУГ НЕРВНО-МЫШЕЧНЫХ ЗАБОЛЕВАНИЙ
Наследственные заболевания
Инфекционные
Воспалительные
Паранеопластические
При соматических заболеваниях
При эндокринных заболеваниях и др.
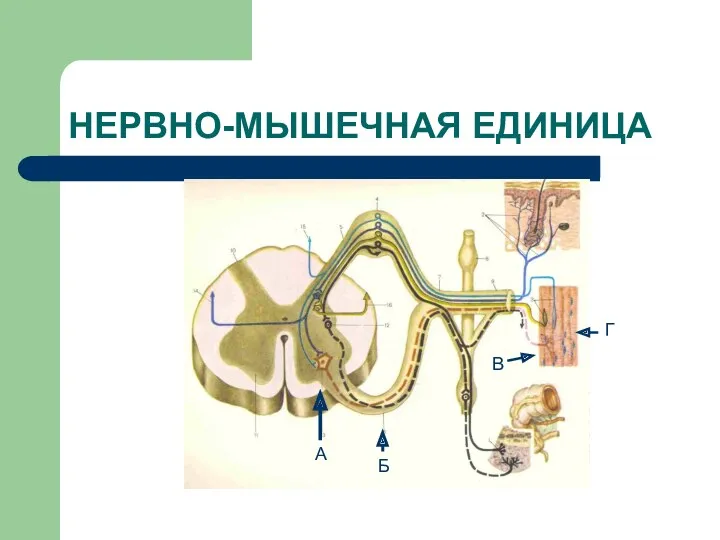
НЕРВНО-МЫШЕЧНАЯ ЕДИНИЦА
А
Б
В
Г
НЕРВНО-МЫШЕЧНАЯ ЕДИНИЦА
А
Б
В
Г

КЛАССИФИКАЦИЯ
А Заболевания, связанные с поражением
передних рогов спинного мозга
Б Болезни, связанные
КЛАССИФИКАЦИЯ
А Заболевания, связанные с поражением
передних рогов спинного мозга
Б Болезни, связанные

ПОРАЖЕНИЕ ПЕРЕДНЕГО РОГА
Только двигательные нарушения
+ фасцикуляции
Отсутствуют чувствительные нарушения
ПОРАЖЕНИЕ ПЕРЕДНЕГО РОГА
Только двигательные нарушения
+ фасцикуляции
Отсутствуют чувствительные нарушения

ПОРАЖЕНИЕ ПЕРИФЕРИЧЕСКИХ НЕРВОВ
Дистальные парезы (кисти, стопы)
В подавляющем большинстве случаев
+ расстройства
ПОРАЖЕНИЕ ПЕРИФЕРИЧЕСКИХ НЕРВОВ
Дистальные парезы (кисти, стопы)
В подавляющем большинстве случаев
+ расстройства

БОЛЕЗНИ СИНАПСА
Патологическая мышечная утомляемость
БОЛЕЗНИ СИНАПСА
Патологическая мышечная утомляемость

МЫШЕЧНОЕ ПОРАЖЕНИЕ
Преимущественное поражение проксимальных отделов конечностей (тазовый, плечевой пояс)
Отсутствуют чувствительные нарушения
МЫШЕЧНОЕ ПОРАЖЕНИЕ
Преимущественное поражение проксимальных отделов конечностей (тазовый, плечевой пояс)
Отсутствуют чувствительные нарушения

ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
ЭМГ
Глобальная
(поверхностные электроды, суммарная активность)
Игольчатая
(активность
ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
ЭМГ
Глобальная
(поверхностные электроды, суммарная активность)
Игольчатая
(активность

ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
Исследование ферментов крови
↑ КФК - креатинфосфокиназа
↑ ЛДГ –
ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
Исследование ферментов крови
↑ КФК - креатинфосфокиназа
↑ ЛДГ –

ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
Биопсия мышц (гистохимическое исследование)
Составление родословных таблиц
ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ
Биопсия мышц (гистохимическое исследование)
Составление родословных таблиц

БОЛЕЗНИ ПЕРЕДНЕГО РОГА
Инфекционные
Клещевой энцефалит
Полиомиелит (болезнь Гейне-Медина)-
эпидемический детский
БОЛЕЗНИ ПЕРЕДНЕГО РОГА
Инфекционные
Клещевой энцефалит
Полиомиелит (болезнь Гейне-Медина)-
эпидемический детский

БОЛЕЗНИ ПЕРЕДНЕГО РОГА
Дегенеративные заболевания
Боковой амиотрофический склероз
(БАС) – болезнь Шарко
дегенерация
БОЛЕЗНИ ПЕРЕДНЕГО РОГА
Дегенеративные заболевания
Боковой амиотрофический склероз
(БАС) – болезнь Шарко
дегенерация
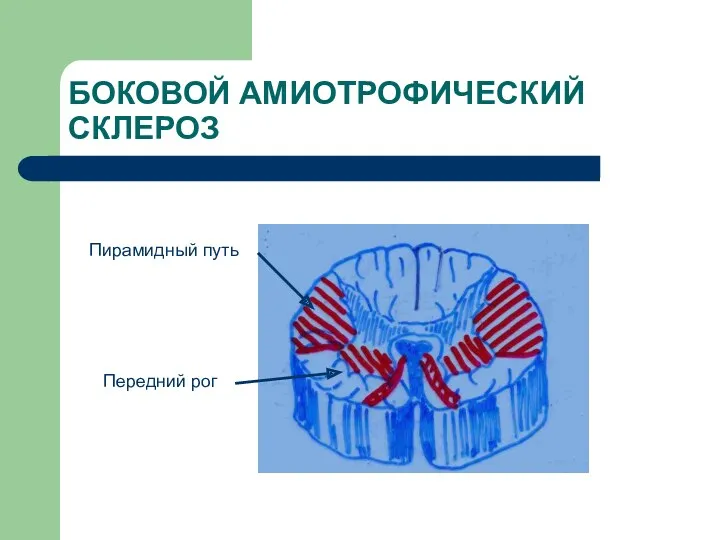
БОКОВОЙ АМИОТРОФИЧЕСКИЙ СКЛЕРОЗ
Пирамидный путь
Передний рог
БОКОВОЙ АМИОТРОФИЧЕСКИЙ СКЛЕРОЗ
Пирамидный путь
Передний рог

УРОВНИ ПОРАЖЕНИЯ ПРИ БАС
Шейное утолщение
Поясничное утолщение
Бульбарный отдел ствола головного мозга
УРОВНИ ПОРАЖЕНИЯ ПРИ БАС
Шейное утолщение
Поясничное утолщение
Бульбарный отдел ствола головного мозга

КЛИНИКА БАС
Смешанные парезы в руках и/или ногах:
признаки вовлечения переднего рога
КЛИНИКА БАС
Смешанные парезы в руках и/или ногах:
признаки вовлечения переднего рога

ТЕЧЕНИЕ БАС
Течение хроническое или подострое
Варианты течения:
как правило, восходящее, но может
ТЕЧЕНИЕ БАС
Течение хроническое или подострое
Варианты течения:
как правило, восходящее, но может

НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ - СПИНАЛЬНЫЕ АМИОТРОФИИ
Ранняя детская форма Вернига-Гофманна
Юношеская форма Кугельберга-Веландер
Бульбо-спинальная амиотрофия
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ - СПИНАЛЬНЫЕ АМИОТРОФИИ
Ранняя детская форма Вернига-Гофманна
Юношеская форма Кугельберга-Веландер
Бульбо-спинальная амиотрофия

РАННЯЯ ДЕТСКАЯ ФОРМА
ВЕРНИГА - ГОФМАННА
Описана в 1891 году
Острая злокачественная инфантильная
РАННЯЯ ДЕТСКАЯ ФОРМА
ВЕРНИГА - ГОФМАННА
Описана в 1891 году
Острая злокачественная инфантильная

КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
Период внутриутробного развития – отсутствие или слабое шевеление
КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
Период внутриутробного развития – отсутствие или слабое шевеление
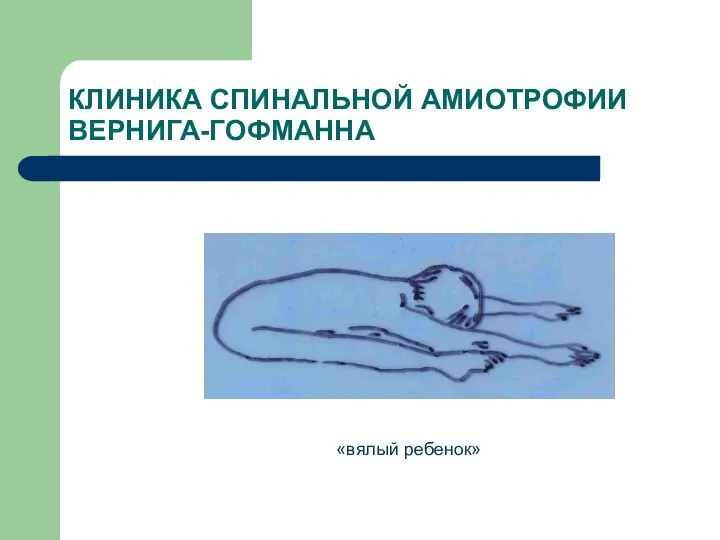
КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
«вялый ребенок»
КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
«вялый ребенок»

КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
Мышечная гипотония
КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ ВЕРНИГА-ГОФМАННА
Мышечная гипотония

ЮНОШЕСКАЯ ФОРМА
КУГЕЛЬБЕРГА - ВЕЛАНДЕР
Описана в 1956 году
Течение доброкачественное
Тип наследования –
ЮНОШЕСКАЯ ФОРМА
КУГЕЛЬБЕРГА - ВЕЛАНДЕР
Описана в 1956 году
Течение доброкачественное
Тип наследования –

КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ КУГЕЛЬБЕРГА-ВЕЛАНДЕР
Начало заболевания в 2 -15 лет (в среднем
КЛИНИКА СПИНАЛЬНОЙ АМИОТРОФИИ КУГЕЛЬБЕРГА-ВЕЛАНДЕР
Начало заболевания в 2 -15 лет (в среднем

СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ
Тяжелая атрофия
мышц плечевого пояса
СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ
Тяжелая атрофия
мышц плечевого пояса

БУЛЬБО-СПИНАЛЬНАЯ АМИОТРОФИЯ КЕННЕДИ
Описана в 1968 году
Доброкачественное течение
Тип наследования – рецессивный, сцепленный
БУЛЬБО-СПИНАЛЬНАЯ АМИОТРОФИЯ КЕННЕДИ
Описана в 1968 году
Доброкачественное течение
Тип наследования – рецессивный, сцепленный
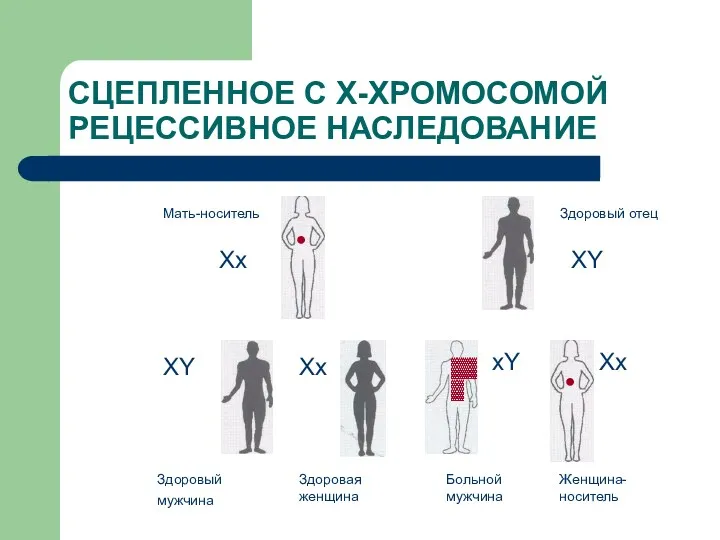
СЦЕПЛЕННОЕ С Х-ХРОМОСОМОЙ РЕЦЕССИВНОЕ НАСЛЕДОВАНИЕ
Мать-носитель
Здоровый отец
Здоровый мужчина
Здоровая женщина
Больной мужчина
Женщина-носитель
СЦЕПЛЕННОЕ С Х-ХРОМОСОМОЙ РЕЦЕССИВНОЕ НАСЛЕДОВАНИЕ
Мать-носитель
Здоровый отец
Здоровый мужчина
Здоровая женщина
Больной мужчина
Женщина-носитель

КЛИНИКА БУЛЬБО-СПИНАЛЬНОЙ АМИОТРОФИИ КЕННЕДИ
Начало в зрелом возрасте (4-я декада)
Проксимальная слабость в
КЛИНИКА БУЛЬБО-СПИНАЛЬНОЙ АМИОТРОФИИ КЕННЕДИ
Начало в зрелом возрасте (4-я декада)
Проксимальная слабость в

БОЛЕЗНИ, СВЯЗАННЫЕ
С ПОРАЖЕНИЕМ ПЕРИФЕРИЧЕСКИХ НЕРВОВ
Общие признаки:
периферические парезы
полиневритический тип нарушения
БОЛЕЗНИ, СВЯЗАННЫЕ
С ПОРАЖЕНИЕМ ПЕРИФЕРИЧЕСКИХ НЕРВОВ
Общие признаки:
периферические парезы
полиневритический тип нарушения

ВИДЫ ПОЛИНЕЙРОПАТИЙ
Аксональные полинейропатии:
при дефиците тиамина, рибофлавина
при отравлении мышьяком
лекарственные
ВИДЫ ПОЛИНЕЙРОПАТИЙ
Аксональные полинейропатии:
при дефиците тиамина, рибофлавина
при отравлении мышьяком
лекарственные

НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ
Группа наследственных сенсомоторных полинейропатий
Описана Ж.Шарко, П.Мари в 1886 г.
НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ
Группа наследственных сенсомоторных полинейропатий
Описана Ж.Шарко, П.Мари в 1886 г.

КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ
Начало с ног:
постепенно нарастает слабость и
КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ
Начало с ног:
постепенно нарастает слабость и
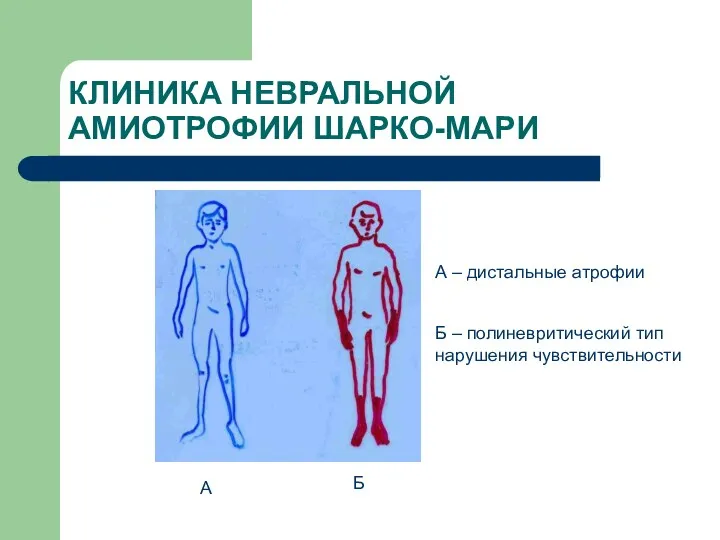
КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ
А
Б
А – дистальные атрофии
Б – полиневритический
КЛИНИКА НЕВРАЛЬНОЙ АМИОТРОФИИ ШАРКО-МАРИ
А
Б
А – дистальные атрофии
Б – полиневритический

Атрофия мышц кисти,
«когтистая лапа»
Невральная амиотрофия Шарко-Мари
Атрофия мышц кисти,
«когтистая лапа»
Невральная амиотрофия Шарко-Мари
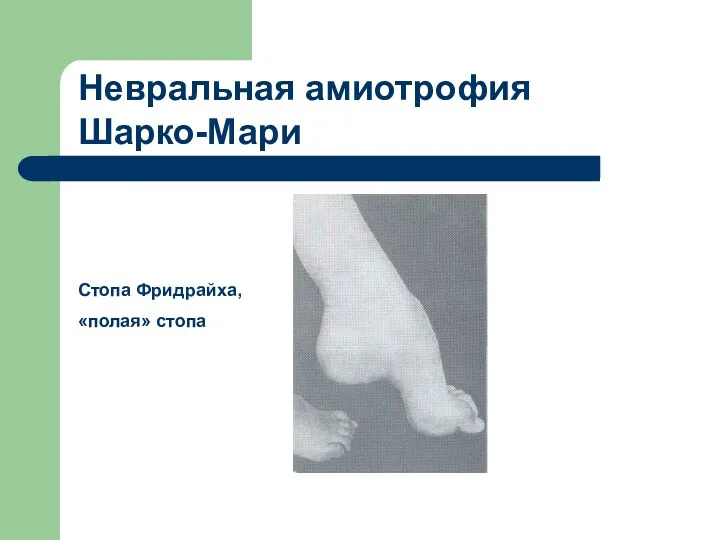
Стопа Фридрайха,
«полая» стопа
Невральная амиотрофия Шарко-Мари
Стопа Фридрайха,
«полая» стопа
Невральная амиотрофия Шарко-Мари

НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ
Типы наследования:
Аутосомно-доминантный
Аутосомно-рецессивный
Сцепленный с Х-хромосомой
НЕВРАЛЬНАЯ АМИОТРОФИЯ ШАРКО-МАРИ
Типы наследования:
Аутосомно-доминантный
Аутосомно-рецессивный
Сцепленный с Х-хромосомой

ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Синдром патологической мышечной утомляемости – нарастание пареза (слабости) к
ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Синдром патологической мышечной утомляемости – нарастание пареза (слабости) к

ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Ботулизм
Миастения
Миастенические синдромы
ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Ботулизм
Миастения
Миастенические синдромы

ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Ботулизм
возникает преходящая блокада пресинаптических структур, нарушается холинергическая трансмиссия
связан с
ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Ботулизм
возникает преходящая блокада пресинаптических структур, нарушается холинергическая трансмиссия
связан с

ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Миастения (Myasthenia gravis)
Заболевание аутоиммунной природы
Вырабатываются антитела к белку ацетилхолиновых
ПОРАЖЕНИЕ НЕРВНО-МЫШЕЧНОГО СИНАПСА
Миастения (Myasthenia gravis)
Заболевание аутоиммунной природы
Вырабатываются антитела к белку ацетилхолиновых

КЛИНИКА МИАСТЕНИИ
Начало, как правило, с глазных мышц (птоз, двоение, сонное выражение
КЛИНИКА МИАСТЕНИИ
Начало, как правило, с глазных мышц (птоз, двоение, сонное выражение

КЛИНИЧЕСКИЕ ПРОБЫ, ВЫЯВЛЯЮЩИЕ ПАТОЛОГИЧЕСКУЮ МЫШЕЧНУЮ УТОМЛЯЕМОСТЬ
Фиксировать взор вверх – 30 секунд
КЛИНИЧЕСКИЕ ПРОБЫ, ВЫЯВЛЯЮЩИЕ ПАТОЛОГИЧЕСКУЮ МЫШЕЧНУЮ УТОМЛЯЕМОСТЬ
Фиксировать взор вверх – 30 секунд

ТЕЧЕНИЕ МИАСТЕНИИ
Характерно течение с ремиссиями
В 20% наблюдаются миастенические кризы (генерализованная мышечная
ТЕЧЕНИЕ МИАСТЕНИИ
Характерно течение с ремиссиями
В 20% наблюдаются миастенические кризы (генерализованная мышечная

ЛЕЧЕНИЕ МИАСТЕНИИ
Антихолинэстеразные
прозерин – быстрого действия
калимин – медленного
Преднизолон (больным старше 50
ЛЕЧЕНИЕ МИАСТЕНИИ
Антихолинэстеразные
прозерин – быстрого действия
калимин – медленного
Преднизолон (больным старше 50

МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Синдром Ламберта-Итона –
паранеопластический синдром
Наблюдается у мужчин старше 40 лет
МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Синдром Ламберта-Итона –
паранеопластический синдром
Наблюдается у мужчин старше 40 лет

МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Клиника
не страдают глазные мышцы
нет реакции на антихолинэстеразные препараты
миастенический синдром может
МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Клиника
не страдают глазные мышцы
нет реакции на антихолинэстеразные препараты
миастенический синдром может

МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Другие причины
Заболевания щитовидной железы (аутоиммунные)
Интоксикация лекарственными препаратами:
неомицин
гентамицин
Д-пенициламин
МИАСТЕНИЧЕСКИЕ СИНДРОМЫ
Другие причины
Заболевания щитовидной железы (аутоиммунные)
Интоксикация лекарственными препаратами:
неомицин
гентамицин
Д-пенициламин

БОЛЕЗНИ, СВЯЗАННЫЕ С ПОРАЖЕНИЕМ МЫШЦ (МИОПАТИИ)
Общие признаки:
Отсутствие чувствительных и вегетативных нарушений
Поражение
БОЛЕЗНИ, СВЯЗАННЫЕ С ПОРАЖЕНИЕМ МЫШЦ (МИОПАТИИ)
Общие признаки:
Отсутствие чувствительных и вегетативных нарушений
Поражение

ПОЛИМИОЗИТ
Не наследственное (аутоиммунное) заболевание
Нарушение клеточного и гуморального иммунитета («воспалительная миопатия»)
Может протекать
ПОЛИМИОЗИТ
Не наследственное (аутоиммунное) заболевание
Нарушение клеточного и гуморального иммунитета («воспалительная миопатия»)
Может протекать

КЛИНИКА ПОЛИМИОЗИТА
Начало – острое или подострое
Мышечная слабость
Недомогание, артралгии, миалгии
Повышение температуры тела
Поражение
КЛИНИКА ПОЛИМИОЗИТА
Начало – острое или подострое
Мышечная слабость
Недомогание, артралгии, миалгии
Повышение температуры тела
Поражение

ПОЛИМИОЗИТ
Данные дополнительного исследования:
Уровень КФК не коррелирует с тяжестью
↑ миоглобин в сыворотке
ПОЛИМИОЗИТ
Данные дополнительного исследования:
Уровень КФК не коррелирует с тяжестью
↑ миоглобин в сыворотке

ВОСПАЛИТЕЛЬНАЯ МИОПАТИЯ (ПОЛИМИОЗИТ)
I
Биопсия мышцы
Воспалительные изменения в мышце
ВОСПАЛИТЕЛЬНАЯ МИОПАТИЯ (ПОЛИМИОЗИТ)
I
Биопсия мышцы
Воспалительные изменения в мышце

НАСЛЕДСТВЕННЫЕ МИОДИСТРОФИИ (МИОПАТИИ)
Известно много форм и вариантов
Псевдогипертрофическая миодистрофия
Дюшена
Миодистрофия
НАСЛЕДСТВЕННЫЕ МИОДИСТРОФИИ (МИОПАТИИ)
Известно много форм и вариантов
Псевдогипертрофическая миодистрофия
Дюшена
Миодистрофия

Неравномерность диаметра мышечных волокон, разрастание соединительной и жировой тканей
ПЕРВИЧНЫЕ
Неравномерность диаметра мышечных волокон, разрастание соединительной и жировой тканей
ПЕРВИЧНЫЕ

ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МИОДИСТРОФИЯ ДЮШЕНА
Описана в 1861 году Дюшеном
В 1879 году Говерс обобщил
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МИОДИСТРОФИЯ ДЮШЕНА
Описана в 1861 году Дюшеном
В 1879 году Говерс обобщил

КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
Первые признаки появляются с момента начала ходьбы (близко
КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
Первые признаки появляются с момента начала ходьбы (близко

КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
Умственная отсталость – 30%.
ЭМГ – признаки первично-мышечного поражения.
Биопсия
КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
Умственная отсталость – 30%.
ЭМГ – признаки первично-мышечного поражения.
Биопсия
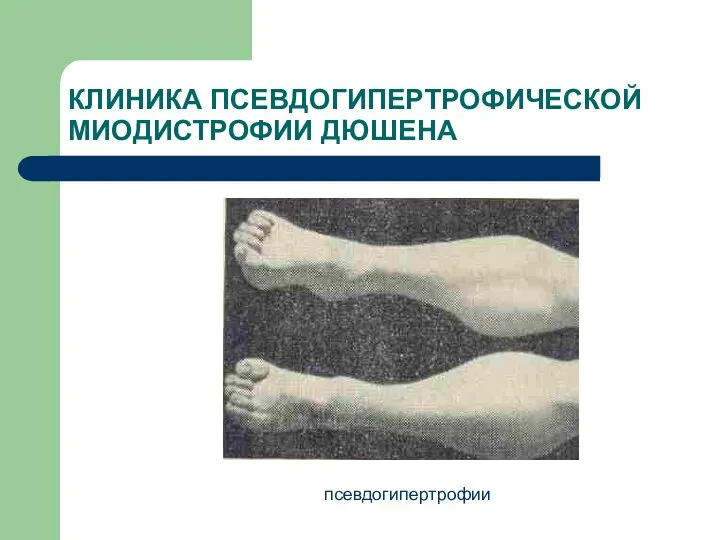
КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
псевдогипертрофии
КЛИНИКА ПСЕВДОГИПЕРТРОФИЧЕСКОЙ МИОДИСТРОФИИ ДЮШЕНА
псевдогипертрофии

Мальчик 5 лет
Наблюдаются псевдогипертрофии мышц, лордоз
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ ФОРМА ДЮШЕНА
Мальчик 5 лет
Наблюдаются псевдогипертрофии мышц, лордоз
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ ФОРМА ДЮШЕНА

СИМПТОМ «ВСТАВАНИЯ ЛЕСЕНКОЙ» ПРИ МИОПАТИИ
СИМПТОМ «ВСТАВАНИЯ ЛЕСЕНКОЙ» ПРИ МИОПАТИИ

МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА
Описана в 1884 -1886 г.г. (Дежерин, Ландузи)
Частота встречаемости–0,4 на 100
МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА
Описана в 1884 -1886 г.г. (Дежерин, Ландузи)
Частота встречаемости–0,4 на 100

КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Дебют в 15-25 лет (≈20 лет)
Слабость и атрофии мышц
КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Дебют в 15-25 лет (≈20 лет)
Слабость и атрофии мышц

КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Слабость мышц лица:
глазные щели не смыкаются ночью
больные не
КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Слабость мышц лица:
глазные щели не смыкаются ночью
больные не

КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Атрофия и слабость мышц плечевого и тазового поясов:
крыловидные лопатки,
КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Атрофия и слабость мышц плечевого и тазового поясов:
крыловидные лопатки,

КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Интеллект не страдает
КФК↑ у 50 - 80%
ЛДГ↑ у 20%
Альдолаза↑
КЛИНИКА МИОДИСТРОФИИ ЛАНДУЗИ-ДЕЖЕРИНА
Интеллект не страдает
КФК↑ у 50 - 80%
ЛДГ↑ у 20%
Альдолаза↑
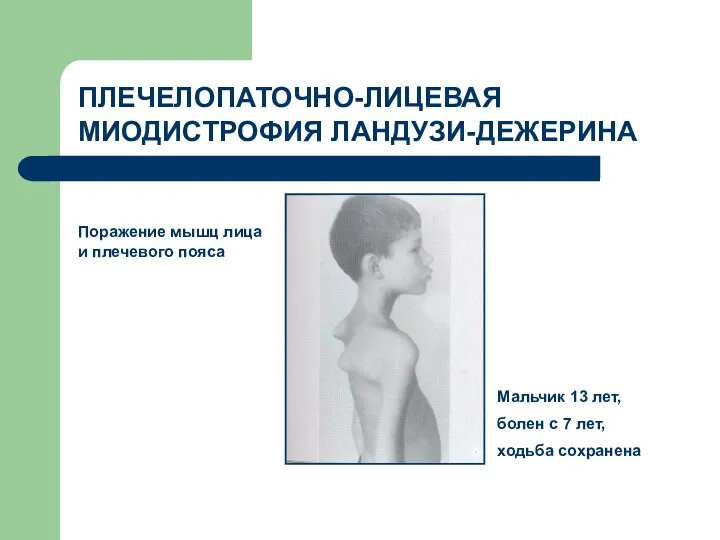
ПЛЕЧЕЛОПАТОЧНО-ЛИЦЕВАЯ МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА
Поражение мышц лица и плечевого пояса
Мальчик 13
ПЛЕЧЕЛОПАТОЧНО-ЛИЦЕВАЯ МИОДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА
Поражение мышц лица и плечевого пояса
Мальчик 13
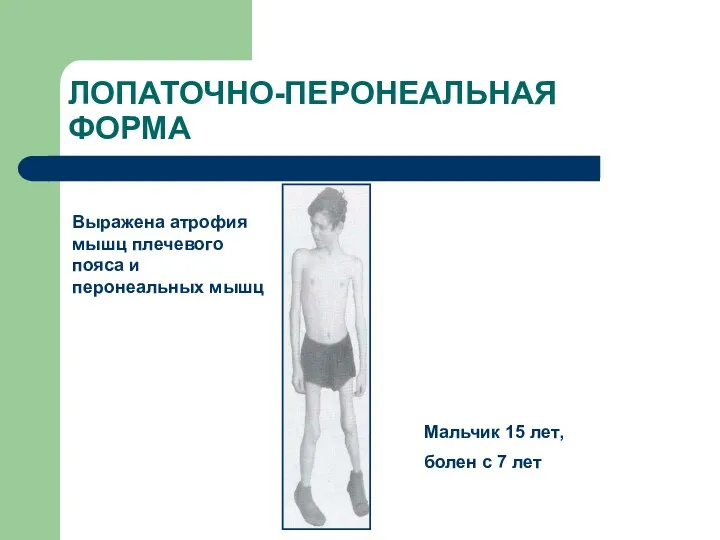
ЛОПАТОЧНО-ПЕРОНЕАЛЬНАЯ ФОРМА
Выражена атрофия мышц плечевого пояса и перонеальных мышц
Мальчик 15
ЛОПАТОЧНО-ПЕРОНЕАЛЬНАЯ ФОРМА
Выражена атрофия мышц плечевого пояса и перонеальных мышц
Мальчик 15

МИОДИСТРОФИЯ ЭРБА-РОТА
Описана в 1884 году Эрбом
Тип наследования аутосомно-рецессивный
Экспрессивность гена у членов
МИОДИСТРОФИЯ ЭРБА-РОТА
Описана в 1884 году Эрбом
Тип наследования аутосомно-рецессивный
Экспрессивность гена у членов

КЛИНИКА МИОДИСТРОФИИ ЭРБА
Дебют во 2-м десятилетии, но может быть и в
КЛИНИКА МИОДИСТРОФИИ ЭРБА
Дебют во 2-м десятилетии, но может быть и в

КЛИНИКА МИОДИСТРОФИИ ЭРБА
Форма Лейдена-Мебиуса
начало с проксимальных отделов ног
Форма Эрба
начало
КЛИНИКА МИОДИСТРОФИИ ЭРБА
Форма Лейдена-Мебиуса
начало с проксимальных отделов ног
Форма Эрба
начало
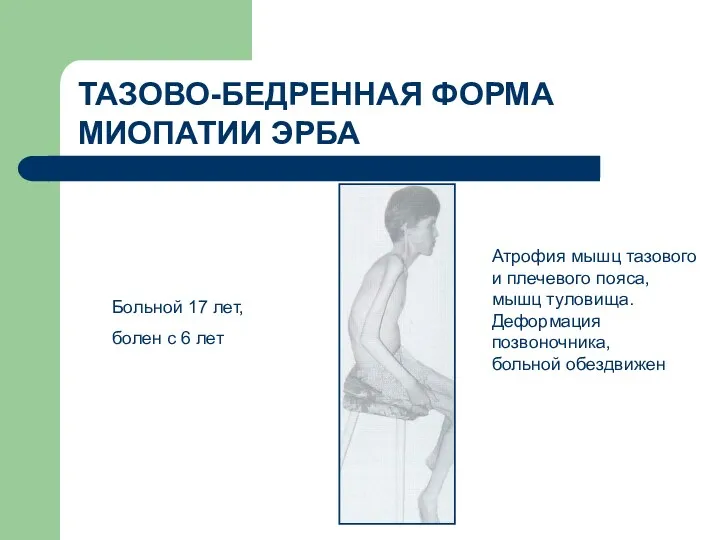
ТАЗОВО-БЕДРЕННАЯ ФОРМА МИОПАТИИ ЭРБА
Больной 17 лет,
болен с 6
ТАЗОВО-БЕДРЕННАЯ ФОРМА МИОПАТИИ ЭРБА
Больной 17 лет,
болен с 6
 Листериялар. Морфология, физиология, листериялар антигені. Экологиясы. Әйелдер патологиясындағы маңызы
Листериялар. Морфология, физиология, листериялар антигені. Экологиясы. Әйелдер патологиясындағы маңызы Prezentatsia_po_biologii_na_temu_Znachenie_pischi_i_eyo_sostav__8_klass
Prezentatsia_po_biologii_na_temu_Znachenie_pischi_i_eyo_sostav__8_klass Внутренняя картина болезни и ее взаимосвязь между психоэмоциональным состоянием у лиц с сахарным диабетом 2 типа
Внутренняя картина болезни и ее взаимосвязь между психоэмоциональным состоянием у лиц с сахарным диабетом 2 типа Лекарственные средства, вызывающие тонические сокращения миометрия матки
Лекарственные средства, вызывающие тонические сокращения миометрия матки Объективный статус при осмотре ребенка
Объективный статус при осмотре ребенка Современные проблемы профилактики ХНИЗ
Современные проблемы профилактики ХНИЗ Ауыз қуыс кілегей қабық ауруларына тағайындалатын дәрілік терпияның салыстырмалы сипаттамасы
Ауыз қуыс кілегей қабық ауруларына тағайындалатын дәрілік терпияның салыстырмалы сипаттамасы Есту қабілеті нашар, көз көруі бұзылған,сөйлеу қабілеті нашар науқастармен қарым-қатынас
Есту қабілеті нашар, көз көруі бұзылған,сөйлеу қабілеті нашар науқастармен қарым-қатынас Исследовательский проект на тему: Соль - вред или польза
Исследовательский проект на тему: Соль - вред или польза Туберкулез органов мочевой системы
Туберкулез органов мочевой системы Міри радіобіологічних ефектів
Міри радіобіологічних ефектів Медицинская статистика. Цели и задачи
Медицинская статистика. Цели и задачи Аномальные маточные кровотечения
Аномальные маточные кровотечения Выпот в полость перикарда
Выпот в полость перикарда Вирус Эбола
Вирус Эбола Классификация шизофрении
Классификация шизофрении Пищеварение. Нарушения экзокринной секреции поджелудочной железы
Пищеварение. Нарушения экзокринной секреции поджелудочной железы Ісіктер туралы жалпы ілім
Ісіктер туралы жалпы ілім Воспалительные заболевания женских половых органов
Воспалительные заболевания женских половых органов Инфекции, передающиеся половым путем
Инфекции, передающиеся половым путем Переливанням крові та донорство
Переливанням крові та донорство Қан физиологиясы
Қан физиологиясы Ерлердің жыныс мүшулурінін даму ақаулары
Ерлердің жыныс мүшулурінін даму ақаулары Помощь при рвоте, кормление тяжело больного пациента
Помощь при рвоте, кормление тяжело больного пациента Психоорганический синдром и когнитивные нарушения – взгляд психиатра
Психоорганический синдром и когнитивные нарушения – взгляд психиатра Морфологические основы почки
Морфологические основы почки Лимфоаденопатии. Дифференциальная диагностика
Лимфоаденопатии. Дифференциальная диагностика Гигиена органов пищеварения. Предупреждение желудочно-кишечных инфекций
Гигиена органов пищеварения. Предупреждение желудочно-кишечных инфекций