- Обмен отдельных аминокислот

Содержание
- 2. Метаболизм глицина, серина, треонина.
- 4. Обмен серосодержащих аминокислот. Цистеин.
- 5. Взаимосвязь обмена серина, глицина, метионина и цистеина:
- 6. Трансметилирование
- 8. Синтез креатина Протекает в 2х органах : почках и печени
- 9. Креатин-Ф играет большую роль особенно для мышц , поскольку поддерживает соотношение АТФ к АДФ в мышцах.
- 10. Обмен одноуглеродных фрагментов.
- 12. Недостаточность фолиевой кислоты. Недостаточность фолиевой кислоты у человека возникает редко. Гиповитаминоз фолиевой кислоты приводит к нарушению
- 13. Фолиевая кислота
- 15. Обмен фенилаланина и тирозина.
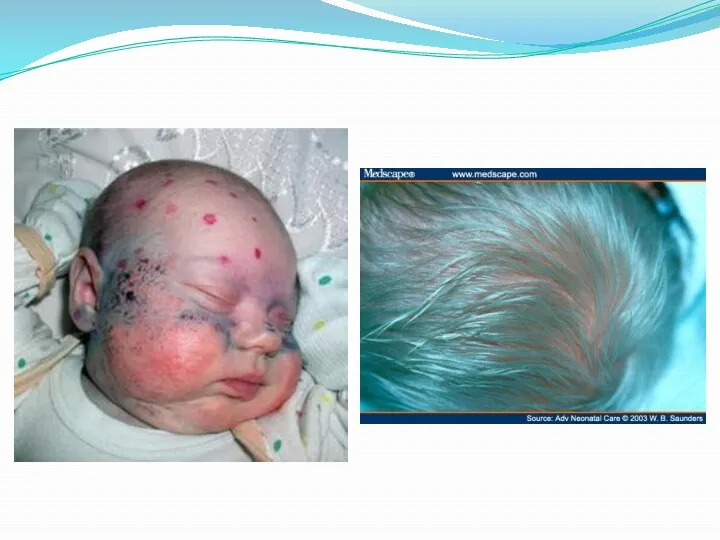
- 17. Фенилкетонурия. Классическая ФКУ - наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы, которые приводят к снижению
- 18. Симптомы фенилкетонурии: Ребенок умственно отсталый, возбудим, своеобразная походка, осанка и поза при сидении, конечности находятся в
- 20. Тирозинемии. Тирозинемия типа I (тирозиноз). Причиной заболевания является, вероятно, дефект фермента фумарилацетоацетатгидролазы, катализирующего расщепление фумарилацетоа-цетата на
- 21. Тирозинемия типа II (синдром Рихнера-Ханхорта). Причина - дефект фермента тирозинаминотрансферазы. Концентрация тирозина в крови больных повышена.
- 22. Поражение кожи при тирозинемии.
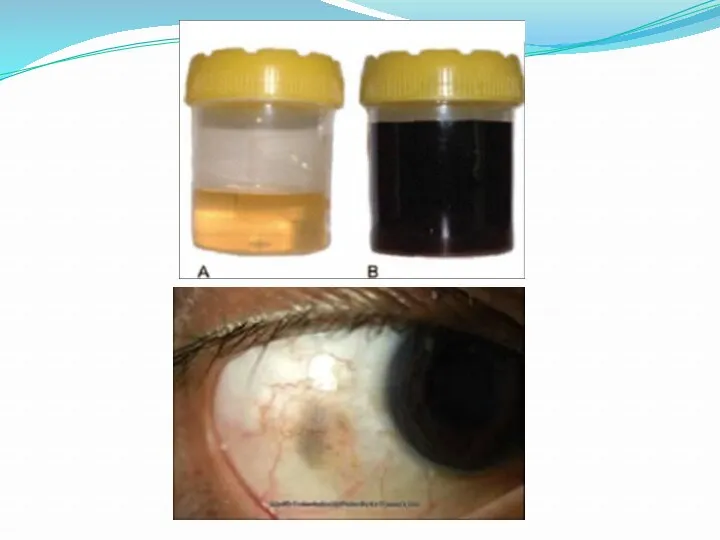
- 23. Алкаптонурия ("чёрная моча") Причина заболевания - дефект диоксигеназы гомогентизиновой кислоты. Для этой болезни характерно выделение с
- 26. Альбинизм. Причина метаболического нарушения - врождённый дефект тирозиназы. Этот фермент катализирует превращение тирозина в ДОФА в
- 28. Болезнь Паркинсона. Заболевание развивается при недостаточности дофамина в чёрной субстанции мозга. При этой патологии снижена активность
- 31. Болезнь мочи кленового сиропа. БМКС вызвана дефицитом комплекса дегидрогеназы альфа-кетокислот с разветвленной цепью, вследствие чего в
- 32. Лейциноз или болезнь мочи кленового сиропа
- 33. Болезнь Вильсона-Коновалова. - врождённое нарушение метаболизма меди, приводящее к тяжелейшим наследственным болезням центральной нервной системы и
- 34. Типичным симптомом болезни является кольцо Кайзера-Флейшера — отложение по периферии роговой оболочки содержащего медь зеленовато-бурого пигмента;
- 37. Скачать презентацию
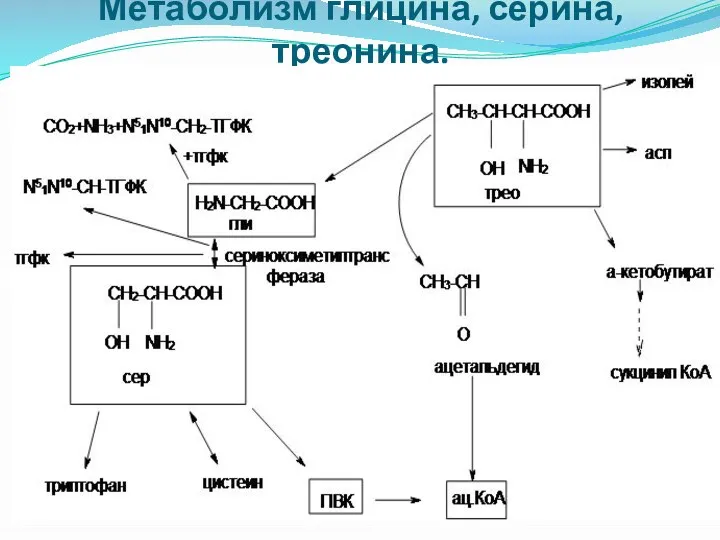
Метаболизм глицина, серина, треонина.
Метаболизм глицина, серина, треонина.
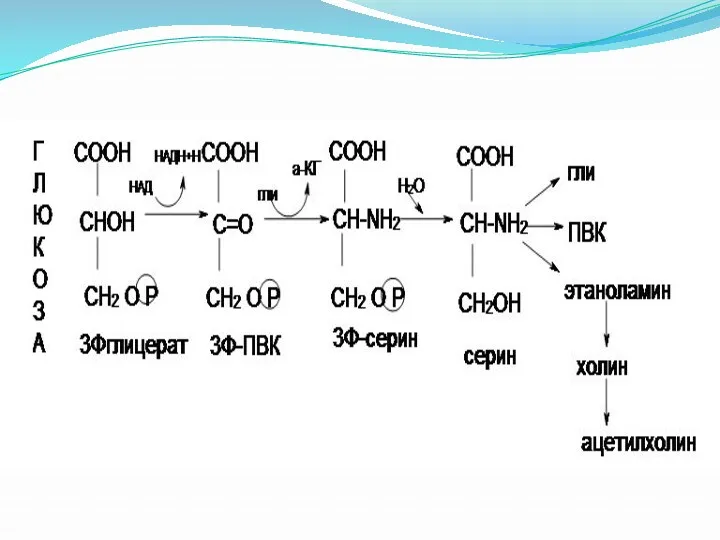
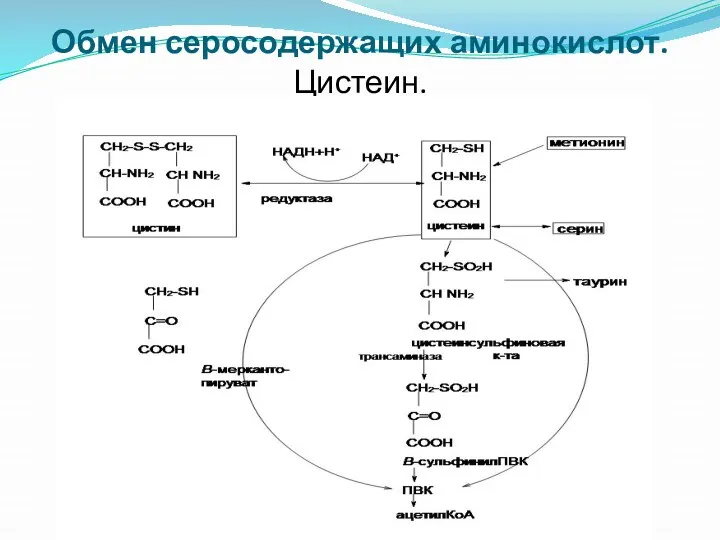
Обмен серосодержащих аминокислот.
Цистеин.
Обмен серосодержащих аминокислот.
Цистеин.
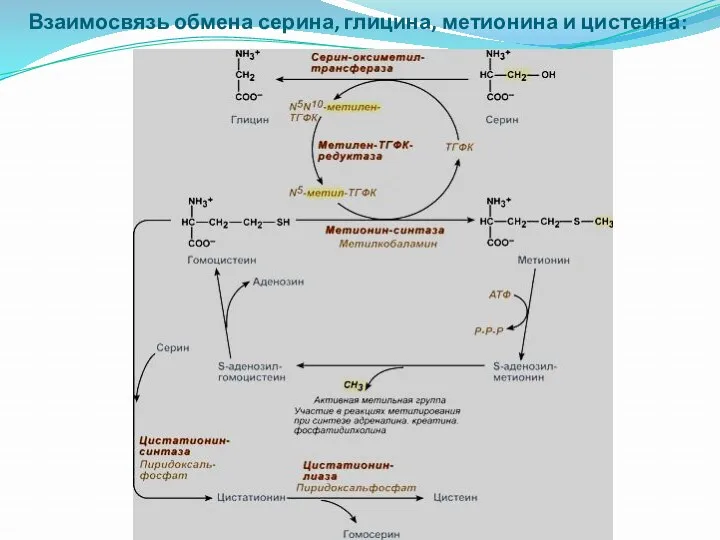
Взаимосвязь обмена серина, глицина, метионина и цистеина:
Взаимосвязь обмена серина, глицина, метионина и цистеина:
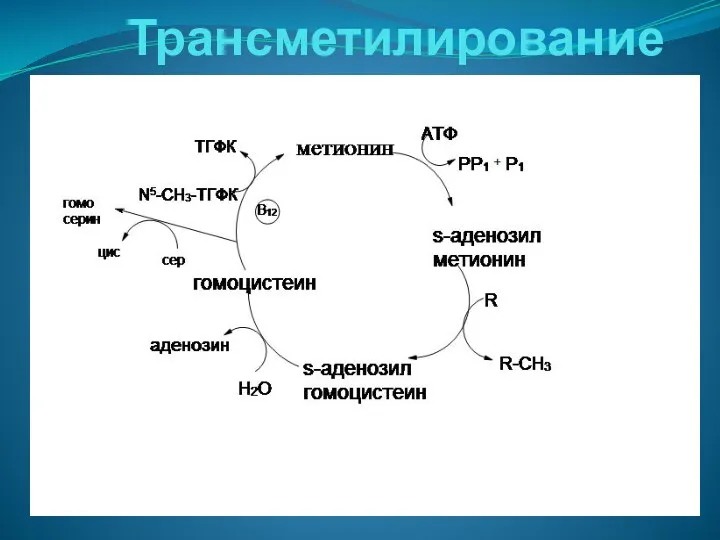
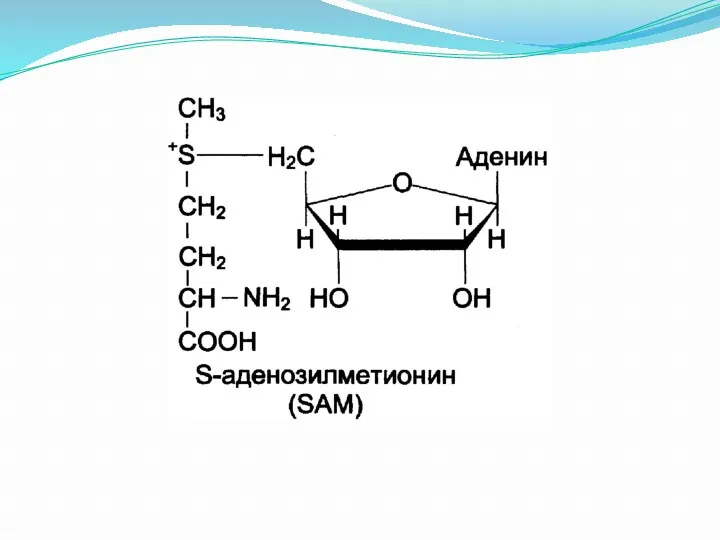
Трансметилирование
Трансметилирование
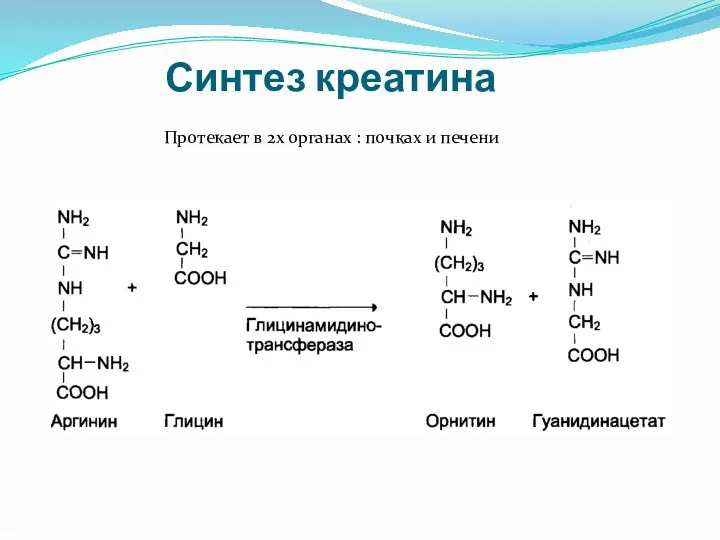
Синтез креатина
Протекает в 2х органах : почках и печени
Синтез креатина
Протекает в 2х органах : почках и печени
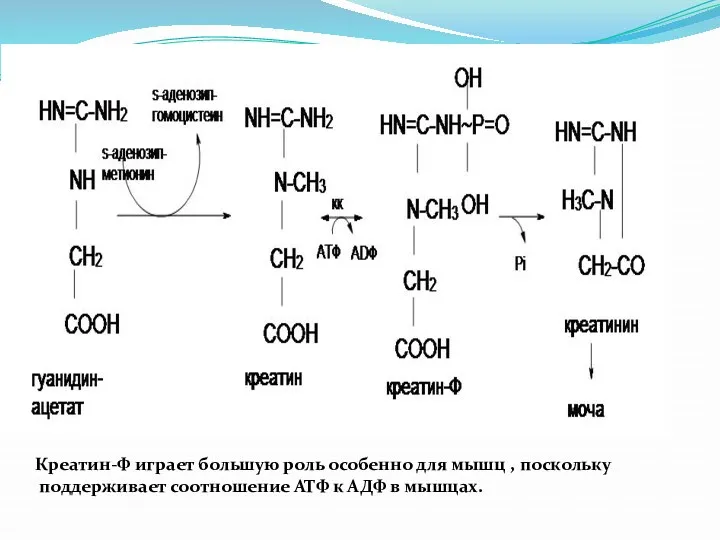
Креатин-Ф играет большую роль особенно для мышц , поскольку
поддерживает соотношение
Креатин-Ф играет большую роль особенно для мышц , поскольку
поддерживает соотношение
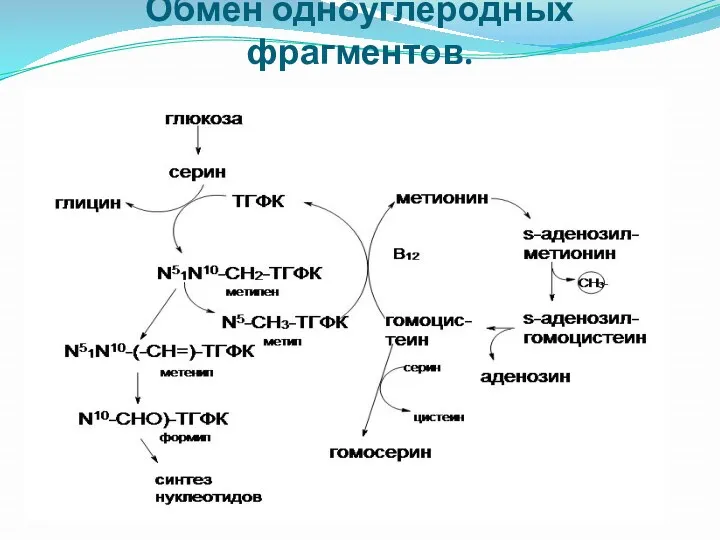
Обмен одноуглеродных фрагментов.
Обмен одноуглеродных фрагментов.

Недостаточность фолиевой кислоты.
Недостаточность фолиевой кислоты у человека возникает редко.
Недостаточность фолиевой кислоты.
Недостаточность фолиевой кислоты у человека возникает редко.
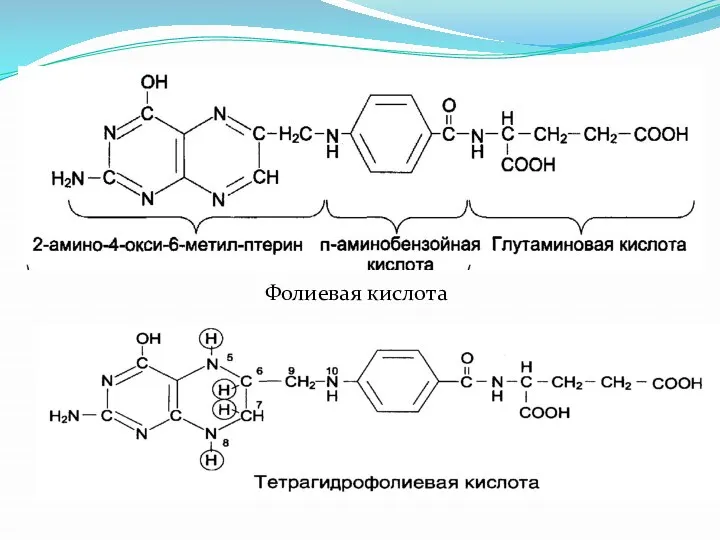
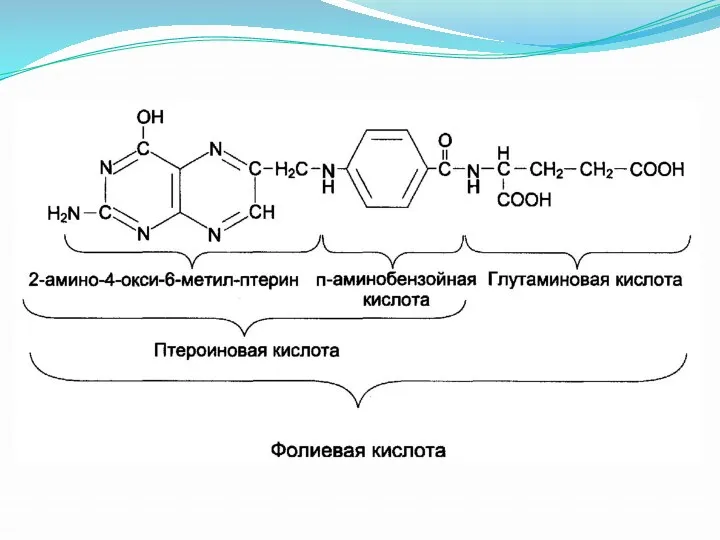
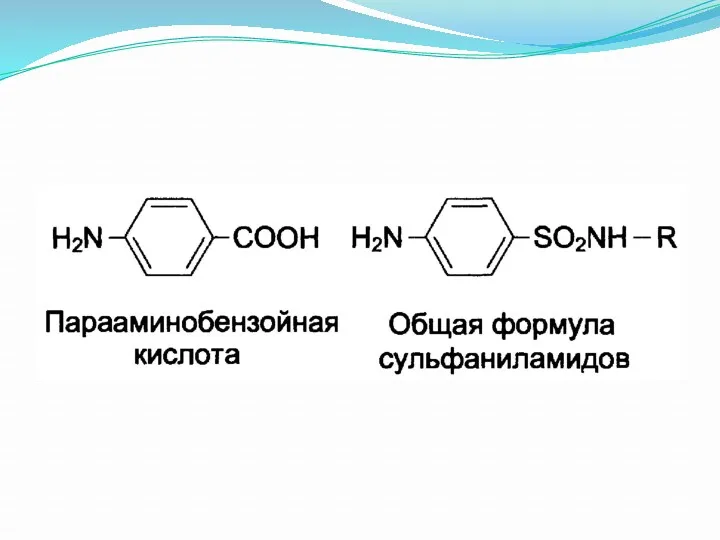
Фолиевая кислота
Фолиевая кислота
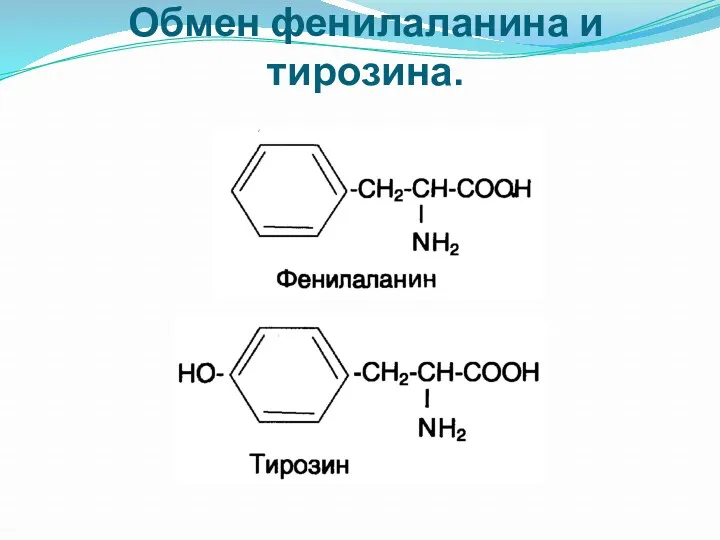
Обмен фенилаланина и тирозина.
Обмен фенилаланина и тирозина.


Фенилкетонурия.
Классическая ФКУ - наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы,
Фенилкетонурия.
Классическая ФКУ - наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы,

Симптомы фенилкетонурии:
Ребенок умственно отсталый, возбудим, своеобразная походка, осанка и
Симптомы фенилкетонурии:
Ребенок умственно отсталый, возбудим, своеобразная походка, осанка и

Тирозинемии.
Тирозинемия типа I (тирозиноз).
Причиной заболевания является, вероятно, дефект фермента фумарилацетоацетатгидролазы,
Тирозинемии.
Тирозинемия типа I (тирозиноз).
Причиной заболевания является, вероятно, дефект фермента фумарилацетоацетатгидролазы,

Тирозинемия типа II (синдром Рихнера-Ханхорта).
Причина - дефект фермента тирозинаминотрансферазы. Концентрация
Тирозинемия типа II (синдром Рихнера-Ханхорта).
Причина - дефект фермента тирозинаминотрансферазы. Концентрация
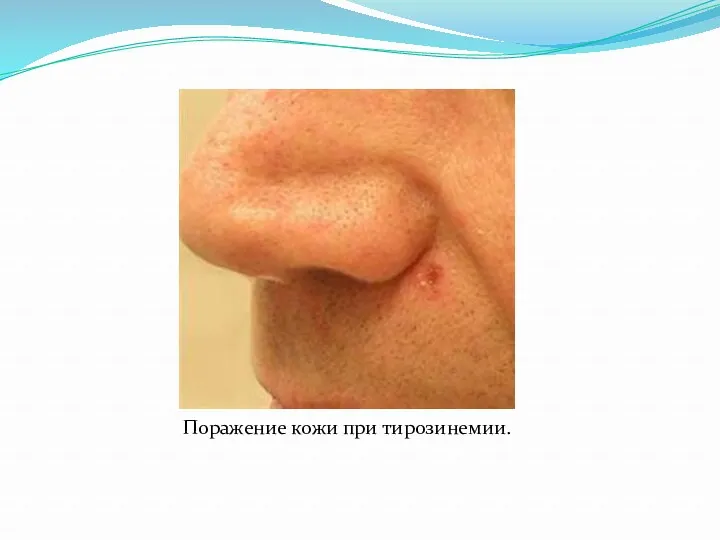
Поражение кожи при тирозинемии.
Поражение кожи при тирозинемии.

Алкаптонурия ("чёрная моча")
Причина заболевания - дефект диоксигеназы гомогентизиновой кислоты. Для этой
Алкаптонурия ("чёрная моча")
Причина заболевания - дефект диоксигеназы гомогентизиновой кислоты. Для этой

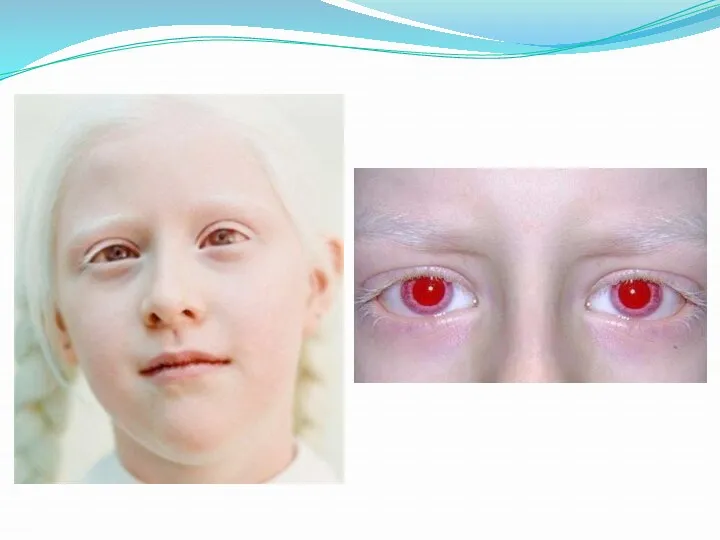
Альбинизм.
Причина метаболического нарушения - врождённый дефект тирозиназы. Этот фермент катализирует превращение
Альбинизм.
Причина метаболического нарушения - врождённый дефект тирозиназы. Этот фермент катализирует превращение

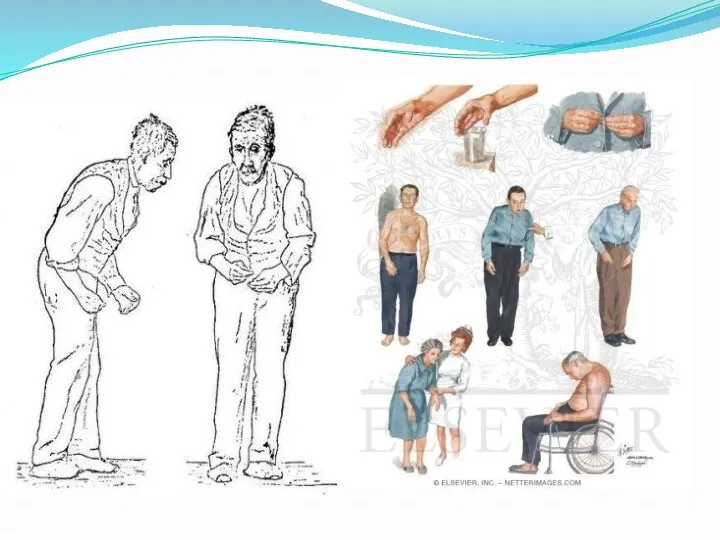
Болезнь Паркинсона.
Заболевание развивается при недостаточности дофамина в чёрной субстанции мозга. При
Болезнь Паркинсона.
Заболевание развивается при недостаточности дофамина в чёрной субстанции мозга. При


Болезнь мочи кленового сиропа.
БМКС вызвана дефицитом комплекса дегидрогеназы альфа-кетокислот
Болезнь мочи кленового сиропа.
БМКС вызвана дефицитом комплекса дегидрогеназы альфа-кетокислот

Лейциноз или болезнь мочи кленового сиропа
Лейциноз или болезнь мочи кленового сиропа

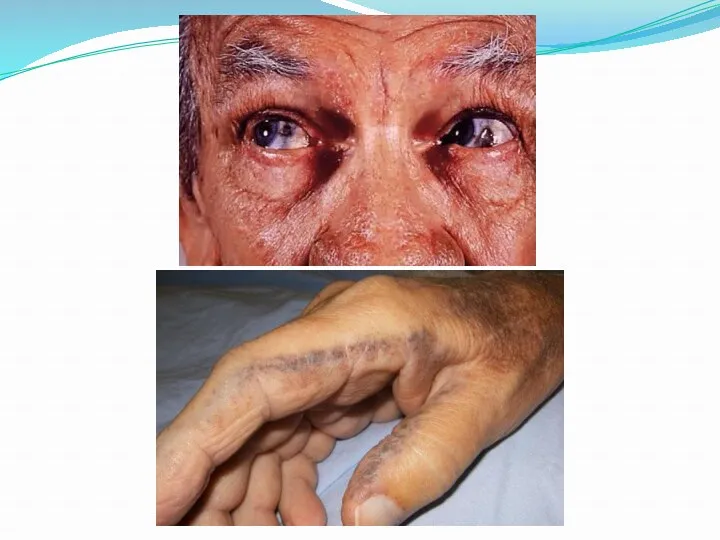
Болезнь Вильсона-Коновалова.
- врождённое нарушение метаболизма меди, приводящее к тяжелейшим наследственным болезням
Болезнь Вильсона-Коновалова.
- врождённое нарушение метаболизма меди, приводящее к тяжелейшим наследственным болезням
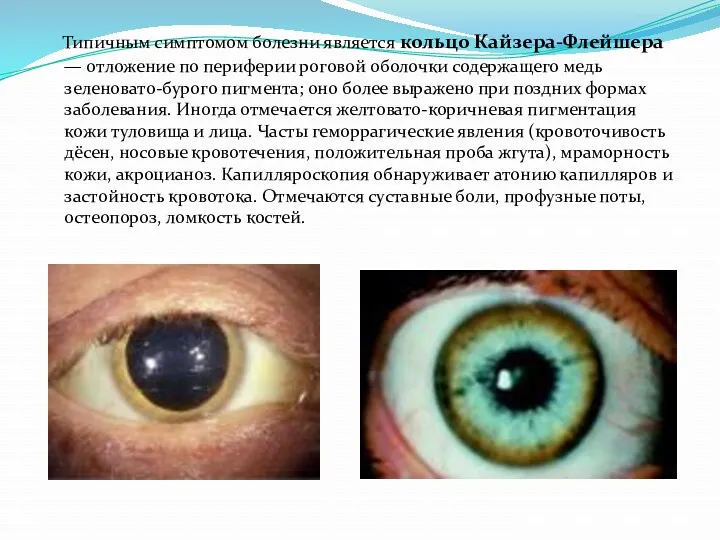
Типичным симптомом болезни является кольцо Кайзера-Флейшера — отложение по периферии
Типичным симптомом болезни является кольцо Кайзера-Флейшера — отложение по периферии
 Психология детей с интеллектуальными нарушениями
Психология детей с интеллектуальными нарушениями Врожденные пороки развития плода
Врожденные пороки развития плода Заболевания печени и желчевыводящих путей
Заболевания печени и желчевыводящих путей Нарушение функции проводимости в электрокардиографии
Нарушение функции проводимости в электрокардиографии ЭКГ. Основы гемодинамики
ЭКГ. Основы гемодинамики Клещи. Болезни, передаваемые клещами, меры защиты. Лекция 15
Клещи. Болезни, передаваемые клещами, меры защиты. Лекция 15 Архітектурні споруди для дітей з фізичними порушеннями
Архітектурні споруди для дітей з фізичними порушеннями Global initiative for chronic obstructive lung disease (GOLD)
Global initiative for chronic obstructive lung disease (GOLD) Хирургиялық науқастарды тамақтандыру
Хирургиялық науқастарды тамақтандыру Заболевания, передающиеся половым путем
Заболевания, передающиеся половым путем Аутоиммунный тиреоидит
Аутоиммунный тиреоидит Переломы. Перелом кости
Переломы. Перелом кости Балалардағы аритмиялар
Балалардағы аритмиялар Сибирская язва. Сибиреязвенный карбункул
Сибирская язва. Сибиреязвенный карбункул Эмоционально-волевая сфера
Эмоционально-волевая сфера Клещевой энцефалит и его профилактика
Клещевой энцефалит и его профилактика Парвавирусный энтерит собак
Парвавирусный энтерит собак Лечение деформирующего артроза
Лечение деформирующего артроза Топография слепой кишки
Топография слепой кишки Cтоп коронавирус
Cтоп коронавирус Иммунодиагностика и иммунотерапия
Иммунодиагностика и иммунотерапия Организация дезинфекции в лечебно-профилактическом учреждении
Организация дезинфекции в лечебно-профилактическом учреждении Хронофармакология
Хронофармакология Искусственное вскармливание детей первого года жизни
Искусственное вскармливание детей первого года жизни Формирование правильной осанки
Формирование правильной осанки Эвтаназия: история проблемы
Эвтаназия: история проблемы Гонорея. Клиническая классификация гонококковой инфекции
Гонорея. Клиническая классификация гонококковой инфекции Проблемы начала человеческой жизни. Семинар 7
Проблемы начала человеческой жизни. Семинар 7