- Паркинсонизм, болезнь Паркинсона и другие экстрапирамидные расстройства

Содержание
- 2. Строение экстрапирамидной системы
- 3. Строение экстрапирамидной системы
- 8. Жан-Мартен Шарко – французский врач-психиатр, специалист по неврологическим болезням, впервые предложивший назвать заболевание болезнью Паркинсона.
- 9. Этиологическая структура паркинсонизма 1. Первичный паркинсонизм Болезнь Паркинсона Ювенильный паркинсонизм 2. Вторичный (симптоматический) паркинсонизм Сосудистый паркинсонизм
- 10. 3. Паркинсонизм при мультисистемных нейродегенеративных заболеваниях ЦНС (паркинсонизм "плюс") 3.1. Преимущественно спорадические формы мультисистемная атрофия прогрессирующий
- 11. 3.2. Наследственные формы болезнь Гентингтона гепатолентикулярная дегенерация спиноцеребеллярные дегенерации семейная кальцификация базальных ганглиев
- 12. Болезнь Паркинсона (идиопатический синдром паркинсонизма, дрожательный паралич) — медленно прогрессирующее хроническое неврологическое заболевание, характерное для лиц
- 13. Возникновение болезни Паркинсона связано с прогрессирующим разрушением и гибелью нейронов, вырабатывающих нейромедиатор дофамин, прежде всего в
- 14. Диагностика паркинсонизма Брадикинезия в сочетании с не менее чем одним из следующих симптомов: 1. мышечная ригидность
- 15. Клинико-диагностические критерии болезни Паркинсона Критерии, подтверждающие диагноз болезни Паркинсона (не менее трех признаков): Асимметричное начало Тремор
- 17. Патоморфология болезни Паркинсона Дегенерация и дептгментация черной субстанции Дегенерация нейронов голубого пятна и покрышки ствола мозга
- 18. Нейрохимические нарушения при паркинсонизме уменьшение синтеза дофамина увеличение количества ацетилхолина увеличение количества глутамата, аспартата уменьшение количества
- 19. Патогенез болезни Паркинсона внешние факторы окислительный стресс увеличение возбуждающих аминокислот деградация белков избыточное накопление ионов кальция
- 20. Стадии болезни Паркинсона (По Хен и Яру) Стадия 0 — нет признаков заболевания. Стадия 1 —
- 21. Особенности поздних стадий болезни Паркинсона Моторные флуктуации Лекарственные дискинезии Акинетические кризы Вегетативные расстройства Когнитивные нарушения
- 22. Лекарственные дискинезии Хореоатетоз мышц конечностей, шеи Оромандибулярная дискинезия Спастическая кривошея Торсионная дистония Дистония конечностей Нарушения позы
- 23. Лечение болезни Паркинсона 1. Фармакотерапия симптоматическая терапия нейропротекторная терапия 2. Медико-социальная реабилитация диспансерное наблюдение школы для
- 24. Противопаркинсонические средства 1. Антихолинергические средства 2. Препараты амантадина 3. ДОФА-содержащие средства 4. Агонисты дофаминовых рецепторов 5.
- 25. Применение агонистов ДА-рецепторов Ранние стадии БП нейропротекция монотерапия комбинация с амантадином, холинолитиком, селегилином Поздние стадии БП
- 26. Агонисты ДА-рецепторов
- 27. Факторы, влияющие на выбор терапии степень тяжести БП возраст эффективность препарата когнитивные нарушения сопутствующие заболевания социальные
- 28. Болезнь Паркинсона Функциональные нарушения Нелекарственные методы Нет Обучение, группа поддержки,ЛФК, питание, психотерапия Фармакотерапия Да Леводопа агонисты
- 29. Комбинированная терапия: Леводопа + Увеличение дозы леводопы Двигательные флюктуации лекарственные дискинезии Неудовлетворительный фармакотерапевтический контроль побочные эффекты
- 30. Болезнь Паркинсона неуклонно прогрессирует. Больные, не получающие лечения, в среднем теряют возможность обслуживать себя самостоятельно через
- 31. При раннем развитии болезни Паркинсона быстрее прогрессируют симптомы нарушения двигательной активности, а при появлении первых симптомов
- 32. Адекватная терапия замедляет развитие ряда симптомов, ведущих к потере трудоспособности больных (мышечной ригидности, гипокинезии, постуральной неустойчивости
- 33. Хорея Гентингтона клинико-диагностические аспекты
- 34. Болезнь Гентингтона — генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30-50 лет и
- 35. Эпидемиология Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3-7:100000, и 1:1000000 среди остальных
- 36. Генетика Ген хантингтин присутствующий у всех людей, кодирует белок хантингтин , расположен на коротком плече 4-й
- 37. Генетика Появление симптомов в различном возрасте. 36-40 повторов приводят к редуцированной пенетрантности формы этого заболевания, которая
- 38. Патогенез Происходит поражение полосатого тела - стриатума, но при прогрессировании заболевания и другие области головного мозга
- 40. Ранние изменения затрагивают Полосатым телом, которое состоит из хвостатого ядра и скорлупы чёрную субстанцию, 3, 5
- 41. Симптомы Симптомы болезни Хантингтона могут проявиться в любом возрасте, но чаще это происходит в 35–44 год.
- 42. Симптомы когнитивные функции : расстройство абстрактного мышления, способности планировать свои действия, оценивать адекватность своих действий, памяти,
- 43. Диагностика Клинические методы Физикальное обследование, иногда в сочетании с психологическим обследованием, позволяет определить область распространения болезни.
- 44. Генетические методы Для проведения генетической диагностики необходим забор крови с определением повторов ЦАГ в каждом НТТаллеле.
- 45. Лечение Тетрабеназин Рекомендованная начальная доза от 12,5 мг от одного до трехраз в день. максимальная допустимая
- 46. Лечение При депрессии - селективные ингибиторы обратного захвата серотонина и миртазапин, Атипичные антипсихотики-при психозах и нарушениях
- 48. Скачать презентацию
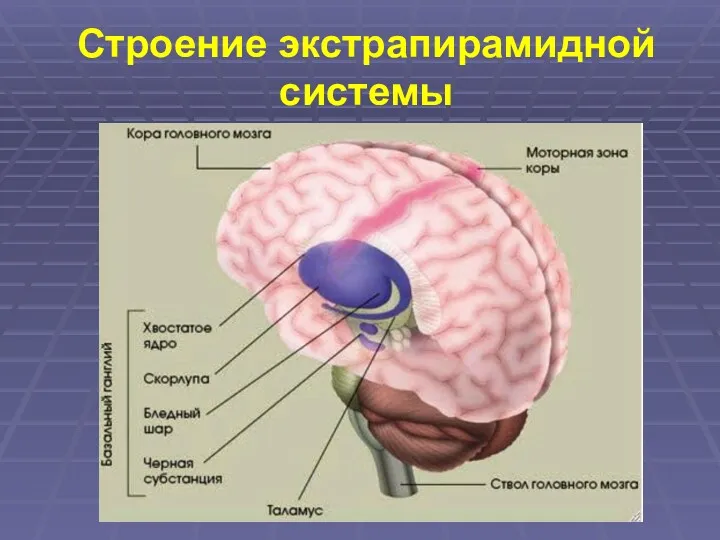
Строение экстрапирамидной системы
Строение экстрапирамидной системы
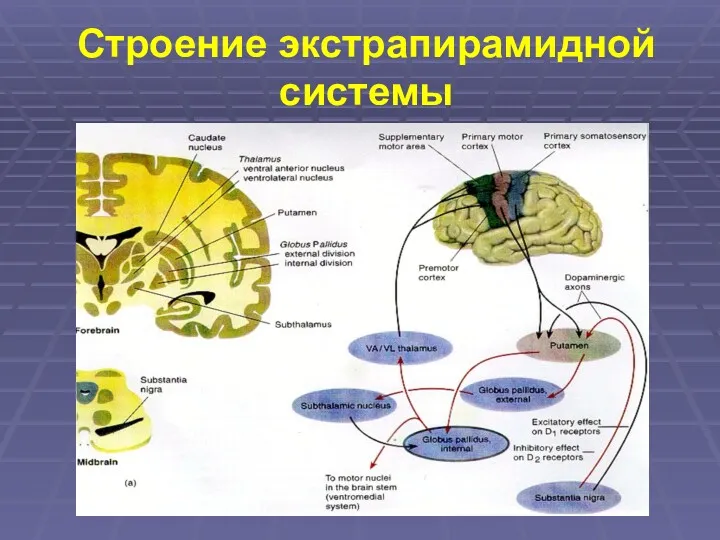
Строение экстрапирамидной системы
Строение экстрапирамидной системы





Жан-Мартен
Шарко – французский врач-психиатр, специалист по неврологическим болезням, впервые предложивший
Жан-Мартен
Шарко – французский врач-психиатр, специалист по неврологическим болезням, впервые предложивший

Этиологическая структура паркинсонизма
1. Первичный паркинсонизм
Болезнь Паркинсона
Ювенильный паркинсонизм
2. Вторичный (симптоматический) паркинсонизм
Этиологическая структура паркинсонизма
1. Первичный паркинсонизм
Болезнь Паркинсона
Ювенильный паркинсонизм
2. Вторичный (симптоматический) паркинсонизм

3. Паркинсонизм при мультисистемных нейродегенеративных заболеваниях ЦНС (паркинсонизм "плюс")
3.1. Преимущественно спорадические
3. Паркинсонизм при мультисистемных нейродегенеративных заболеваниях ЦНС (паркинсонизм "плюс")
3.1. Преимущественно спорадические

3.2. Наследственные формы
болезнь Гентингтона
гепатолентикулярная дегенерация
спиноцеребеллярные дегенерации
семейная кальцификация базальных ганглиев
3.2. Наследственные формы
болезнь Гентингтона
гепатолентикулярная дегенерация
спиноцеребеллярные дегенерации
семейная кальцификация базальных ганглиев

Болезнь Паркинсона
(идиопатический синдром паркинсонизма, дрожательный паралич) — медленно прогрессирующее хроническое неврологическое
Болезнь Паркинсона
(идиопатический синдром паркинсонизма, дрожательный паралич) — медленно прогрессирующее хроническое неврологическое

Возникновение болезни Паркинсона связано с прогрессирующим разрушением и гибелью нейронов, вырабатывающих
Возникновение болезни Паркинсона связано с прогрессирующим разрушением и гибелью нейронов, вырабатывающих

Диагностика паркинсонизма
Брадикинезия в сочетании с не менее чем одним из следующих
Диагностика паркинсонизма
Брадикинезия в сочетании с не менее чем одним из следующих

Клинико-диагностические критерии болезни Паркинсона
Критерии, подтверждающие диагноз болезни Паркинсона (не менее трех
Клинико-диагностические критерии болезни Паркинсона
Критерии, подтверждающие диагноз болезни Паркинсона (не менее трех
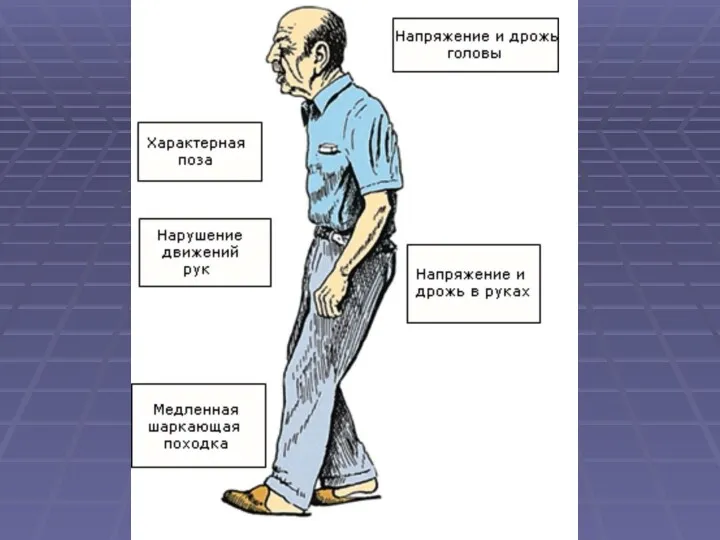

Патоморфология болезни Паркинсона
Дегенерация и дептгментация черной субстанции
Дегенерация нейронов голубого пятна и
Патоморфология болезни Паркинсона
Дегенерация и дептгментация черной субстанции
Дегенерация нейронов голубого пятна и

Нейрохимические нарушения при паркинсонизме
уменьшение синтеза дофамина
увеличение количества ацетилхолина
увеличение количества глутамата, аспартата
уменьшение
Нейрохимические нарушения при паркинсонизме
уменьшение синтеза дофамина
увеличение количества ацетилхолина
увеличение количества глутамата, аспартата
уменьшение
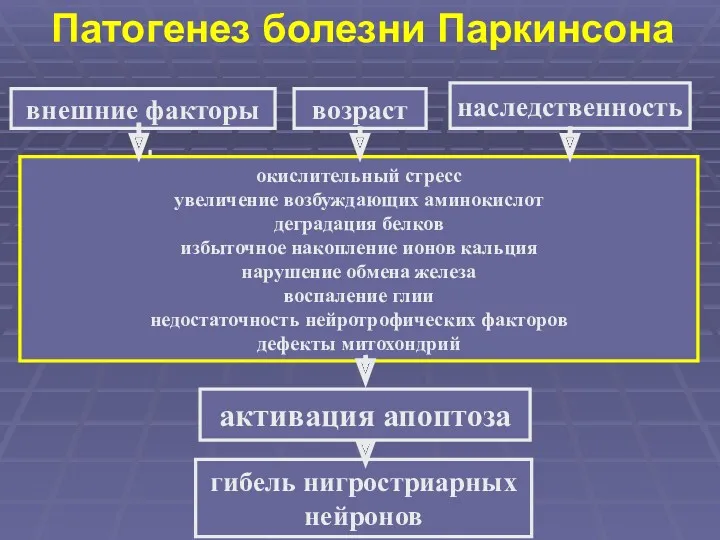
Патогенез болезни Паркинсона
внешние факторы
окислительный стресс
увеличение возбуждающих аминокислот
деградация белков
избыточное накопление ионов
Патогенез болезни Паркинсона
внешние факторы
окислительный стресс
увеличение возбуждающих аминокислот
деградация белков
избыточное накопление ионов

Стадии болезни Паркинсона
(По Хен и Яру)
Стадия 0 — нет признаков
Стадии болезни Паркинсона
(По Хен и Яру)
Стадия 0 — нет признаков

Особенности поздних стадий болезни Паркинсона
Моторные флуктуации
Лекарственные дискинезии
Акинетические кризы
Вегетативные расстройства
Когнитивные нарушения
Особенности поздних стадий болезни Паркинсона
Моторные флуктуации
Лекарственные дискинезии
Акинетические кризы
Вегетативные расстройства
Когнитивные нарушения

Лекарственные дискинезии
Хореоатетоз мышц конечностей, шеи
Оромандибулярная дискинезия
Спастическая кривошея
Торсионная
Лекарственные дискинезии
Хореоатетоз мышц конечностей, шеи
Оромандибулярная дискинезия
Спастическая кривошея
Торсионная

Лечение болезни Паркинсона
1. Фармакотерапия
симптоматическая терапия
нейропротекторная терапия
2. Медико-социальная реабилитация
диспансерное
Лечение болезни Паркинсона
1. Фармакотерапия
симптоматическая терапия
нейропротекторная терапия
2. Медико-социальная реабилитация
диспансерное

Противопаркинсонические средства
1. Антихолинергические средства
2. Препараты амантадина
3. ДОФА-содержащие средства
4. Агонисты дофаминовых
рецепторов
5.
Противопаркинсонические средства
1. Антихолинергические средства
2. Препараты амантадина
3. ДОФА-содержащие средства
4. Агонисты дофаминовых
рецепторов
5.

Применение агонистов ДА-рецепторов
Ранние стадии БП
нейропротекция
монотерапия
комбинация с амантадином,
Применение агонистов ДА-рецепторов
Ранние стадии БП
нейропротекция
монотерапия
комбинация с амантадином,

Агонисты ДА-рецепторов
Агонисты ДА-рецепторов

Факторы, влияющие на выбор терапии
степень тяжести БП
возраст
эффективность препарата
Факторы, влияющие на выбор терапии
степень тяжести БП
возраст
эффективность препарата
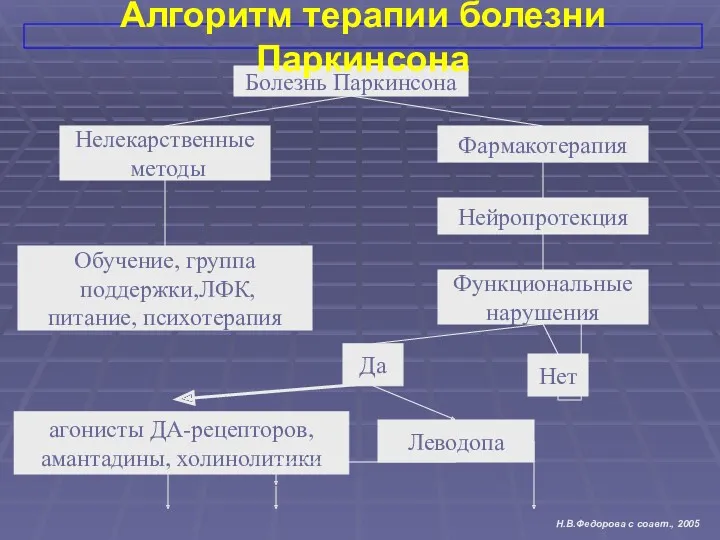
Болезнь Паркинсона
Функциональные
нарушения
Нелекарственные
методы
Нет
Обучение, группа
поддержки,ЛФК,
питание, психотерапия
Фармакотерапия
Да
Леводопа
агонисты ДА-рецепторов,
амантадины, холинолитики
Нейропротекция
Алгоритм терапии болезни Паркинсона
Болезнь Паркинсона
Функциональные
нарушения
Нелекарственные
методы
Нет
Обучение, группа
поддержки,ЛФК,
питание, психотерапия
Фармакотерапия
Да
Леводопа
агонисты ДА-рецепторов,
амантадины, холинолитики
Нейропротекция
Алгоритм терапии болезни Паркинсона
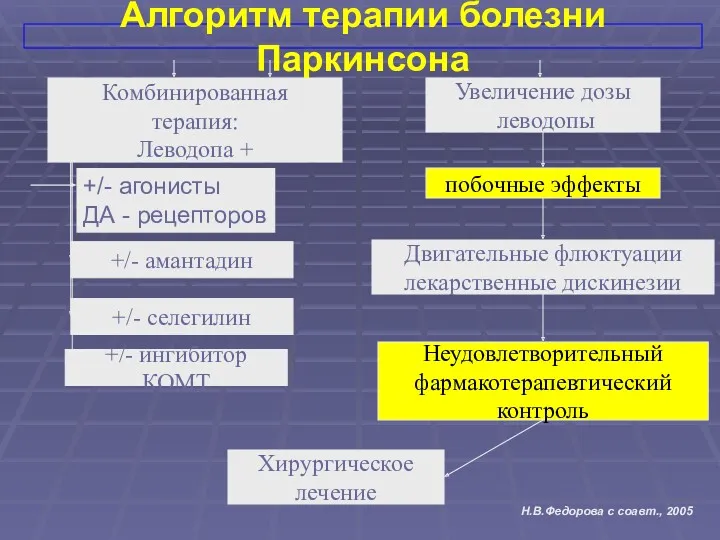
Комбинированная
терапия:
Леводопа +
Увеличение дозы
леводопы
Двигательные флюктуации
лекарственные дискинезии
Неудовлетворительный
фармакотерапевтический
контроль
побочные эффекты
Хирургическое
лечение
+/- селегилин
+/-
Комбинированная
терапия:
Леводопа +
Увеличение дозы
леводопы
Двигательные флюктуации
лекарственные дискинезии
Неудовлетворительный
фармакотерапевтический
контроль
побочные эффекты
Хирургическое
лечение
+/- селегилин
+/-

Болезнь Паркинсона неуклонно прогрессирует.
Больные, не получающие лечения, в среднем теряют
Болезнь Паркинсона неуклонно прогрессирует.
Больные, не получающие лечения, в среднем теряют

При раннем развитии болезни Паркинсона быстрее прогрессируют симптомы нарушения двигательной активности,
При раннем развитии болезни Паркинсона быстрее прогрессируют симптомы нарушения двигательной активности,

Адекватная терапия замедляет развитие ряда симптомов, ведущих к потере трудоспособности больных
Адекватная терапия замедляет развитие ряда симптомов, ведущих к потере трудоспособности больных

Хорея Гентингтона
клинико-диагностические аспекты
Хорея Гентингтона
клинико-диагностические аспекты

Болезнь Гентингтона — генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в
Болезнь Гентингтона — генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в

Эпидемиология
Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3-7:100000,
Эпидемиология Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3-7:100000,

Генетика
Ген хантингтин присутствующий у всех людей, кодирует белок хантингтин , расположен на коротком плече 4-й хромосомы .
Генетика
Ген хантингтин присутствующий у всех людей, кодирует белок хантингтин , расположен на коротком плече 4-й хромосомы .

Генетика
Появление симптомов в различном возрасте. 36-40 повторов приводят к редуцированной пенетрантности формы этого
Генетика
Появление симптомов в различном возрасте. 36-40 повторов приводят к редуцированной пенетрантности формы этого

Патогенез
Происходит поражение полосатого тела - стриатума, но при прогрессировании заболевания и
Патогенез
Происходит поражение полосатого тела - стриатума, но при прогрессировании заболевания и
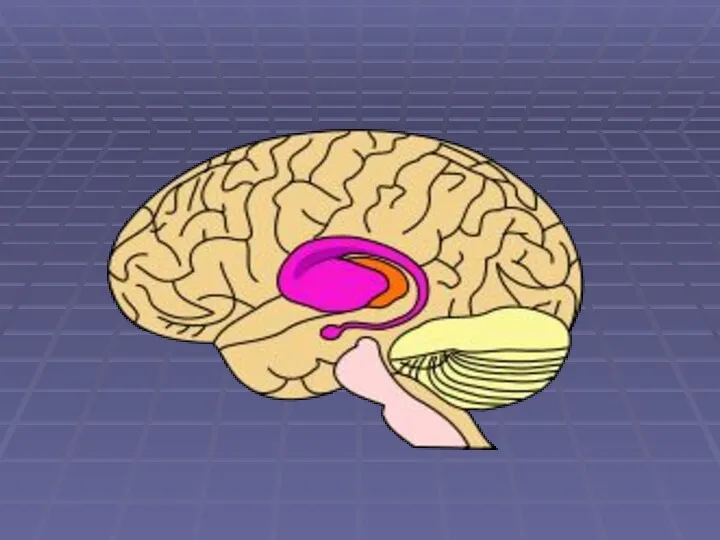

Ранние изменения затрагивают
Полосатым телом, которое состоит из хвостатого ядра и скорлупы
чёрную
Ранние изменения затрагивают
Полосатым телом, которое состоит из хвостатого ядра и скорлупы
чёрную

Симптомы
Симптомы болезни Хантингтона могут проявиться в любом возрасте, но чаще это
Симптомы
Симптомы болезни Хантингтона могут проявиться в любом возрасте, но чаще это

Симптомы
когнитивные функции : расстройство абстрактного мышления, способности планировать свои действия, оценивать
Симптомы
когнитивные функции : расстройство абстрактного мышления, способности планировать свои действия, оценивать

Диагностика
Клинические методы
Физикальное обследование, иногда в сочетании с психологическим обследованием, позволяет определить
Диагностика
Клинические методы
Физикальное обследование, иногда в сочетании с психологическим обследованием, позволяет определить

Генетические методы
Для проведения генетической диагностики необходим забор крови с определением повторов
Генетические методы
Для проведения генетической диагностики необходим забор крови с определением повторов

Лечение
Тетрабеназин Рекомендованная начальная доза от 12,5 мг от одного до трехраз в
Лечение
Тетрабеназин Рекомендованная начальная доза от 12,5 мг от одного до трехраз в

Лечение
При депрессии - селективные ингибиторы обратного захвата серотонина и миртазапин,
Атипичные антипсихотики-при психозах и нарушениях
Лечение
При депрессии - селективные ингибиторы обратного захвата серотонина и миртазапин,
Атипичные антипсихотики-при психозах и нарушениях
 Острый панкреатит
Острый панкреатит Атеросклероз: патогенез, эпидемиология, оценка риска ССЗ. Характеристика дислипидемий. Семейная гиперхолестеринемия
Атеросклероз: патогенез, эпидемиология, оценка риска ССЗ. Характеристика дислипидемий. Семейная гиперхолестеринемия Принципы диспансерного наблюдения детей
Принципы диспансерного наблюдения детей Первая медицинская помощь при травмах
Первая медицинская помощь при травмах Сестринский уход за пациентами с пупочными грыжами
Сестринский уход за пациентами с пупочными грыжами Зомління (непритомність)
Зомління (непритомність) Характеристика нарушений фонематического слуха у детей старшего дошкольного возраста с кохлеарным имплантом
Характеристика нарушений фонематического слуха у детей старшего дошкольного возраста с кохлеарным имплантом Использование КТ для диагностики травм у мелких домашних животных
Использование КТ для диагностики травм у мелких домашних животных Аталық жыныс мүшесі және ұма ағзаларының аномалиялары
Аталық жыныс мүшесі және ұма ағзаларының аномалиялары Жұқпалы аурулардан иммунды әдіспен алдын ала сақтану (Иммунопрофилактика)
Жұқпалы аурулардан иммунды әдіспен алдын ала сақтану (Иммунопрофилактика) ИБС. Стенокардия
ИБС. Стенокардия Инфекциялық аурулардан өлгендерді зерттеу әдістері
Инфекциялық аурулардан өлгендерді зерттеу әдістері Туберкулезный менингит
Туберкулезный менингит Микозы. 235 группа
Микозы. 235 группа Төменгі жақ сүйегінің жазықтықтағы барлық бағыттағы қозғалысын қайталайтын заманауи қондырғылар (артикулятор)
Төменгі жақ сүйегінің жазықтықтағы барлық бағыттағы қозғалысын қайталайтын заманауи қондырғылар (артикулятор) Осмотр новорожденного в родильном зале
Осмотр новорожденного в родильном зале Аневризмы сосудов ГМ
Аневризмы сосудов ГМ Патронаж здорового новорожденного на дому с оформлением учебной амбулаторной карты
Патронаж здорового новорожденного на дому с оформлением учебной амбулаторной карты Дені сау нәрестеге үйде патронаж жасап амбулаториялық оқу картасын толтыру
Дені сау нәрестеге үйде патронаж жасап амбулаториялық оқу картасын толтыру Наркотические анальгетики
Наркотические анальгетики Антигендер жалпы сипаттамасы
Антигендер жалпы сипаттамасы Новые горизонты в лечении ИБС
Новые горизонты в лечении ИБС Травмы мягких тканей
Травмы мягких тканей Вигодовування дітей першого року життя
Вигодовування дітей першого року життя Заболевания сосудов и основы нарушения регионарного кровообращения
Заболевания сосудов и основы нарушения регионарного кровообращения Лекция 13. Психотропные средства-2. Антидепрессанты. Психостимуляторы. Ноотропные. Общетонизирующие средства. Аналептики
Лекция 13. Психотропные средства-2. Антидепрессанты. Психостимуляторы. Ноотропные. Общетонизирующие средства. Аналептики Захворювання нирок та їх профілактика
Захворювання нирок та їх профілактика Психофизиологические основы учебного труда и интеллектуальной деятельности студентов
Психофизиологические основы учебного труда и интеллектуальной деятельности студентов