- Первичные иммунодефициты

Содержание
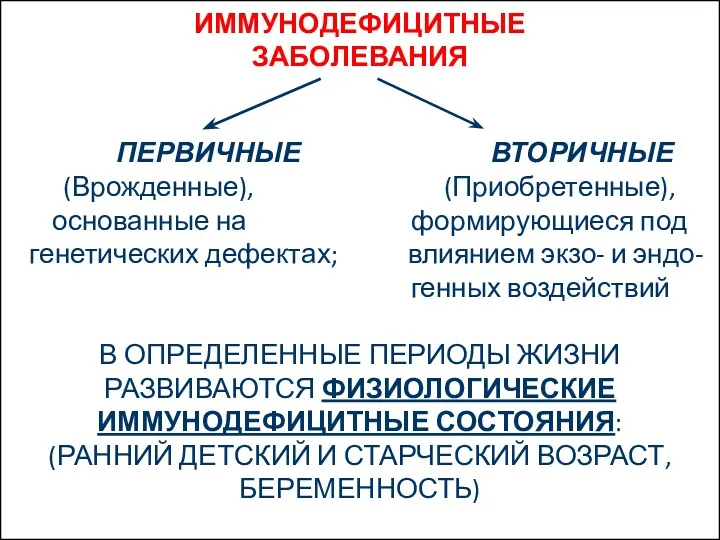
- 2. ИММУНОДЕФИЦИТНЫЕ ЗАБОЛЕВАНИЯ ПЕРВИЧНЫЕ ВТОРИЧНЫЕ (Врожденные), (Приобретенные), основанные на формирующиеся под генетических дефектах; влиянием экзо- и эндо-
- 3. ПЕРВИЧНЫЕ ИММУНОДЕФИЦИТЫ - Заболевания иммунной системы, которые развиваются в результате генетически обусловленного блока клеточных или молекулярных
- 4. Первый случай иммунодефицита описал военный педиатр армии США полковник Ogden Carr BRUTON в 1952 году («Agammaglobulinemia»,
- 5. «ИММУНОДЕФИЦИТЫ НАШИ УЧИТЕЛЯ» «ИММУНОДЕФИЦИТЫ ПРОДОЛЖАЮТ НАС УЧИТЬ» Роберт Алан ГУД (Good) (1922-2003) АМЕРИКАНСКИЙ ПЕДИАТР - ОДИН
- 6. Частота встречаемости ПИД соответствует другим генетическим дефектам человека (1:10000-15000) селективный дефицит IgА (1:300–1:700); ОВИН (1:7000–1:200 000);
- 7. Основные этапы изучения первичных иммунодефицитов Изучение ПИД началось в 50-х годах, когда применение антибиотиков и Ig
- 8. КЛАССИФИКАЦИИ ПЕРВИЧНЫХ ИММУНОДЕФИЦИТОВ (по данным ВОЗ) 1. 1968 – поражение клеточного и гуморального иммунитета, различные варианты
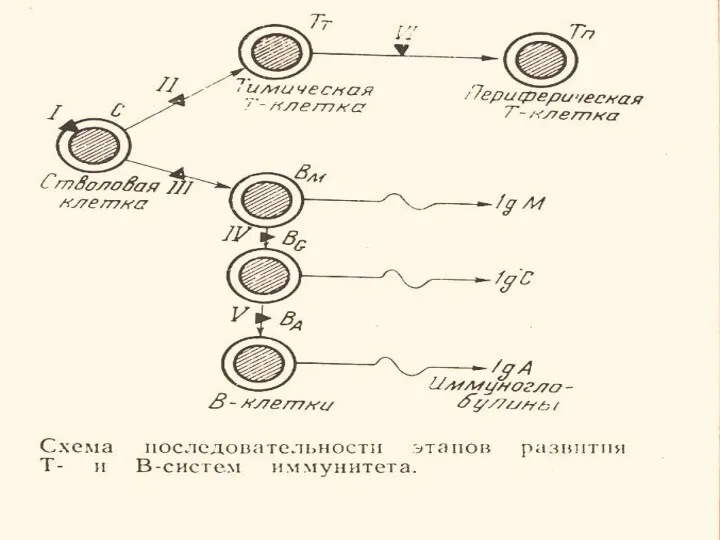
- 9. ОТЕЧЕСТВЕННАЯ КЛАССИФИКАЦИЯ ПЕРВИЧНЫХ ИММУНОДЕФИЦИТОВ ОСНОВАНА НА ГЕНЕТИЧЕСКИХ БЛОКАХ РАЗВИТИЯ Т- И В-КЛЕТОК (по крайней мере 12
- 11. Классификация первичных ИД (2007г.) 1. Комбинированные Т- и В-клеточные иммунодефициты (8,4%); 2. ИД с преимущественным нарушением
- 12. 9. Фенокопии ПИД Наследуемые иммунодефициты, не связанные с мутациями в генах зародышевой линии, возникают по механизму
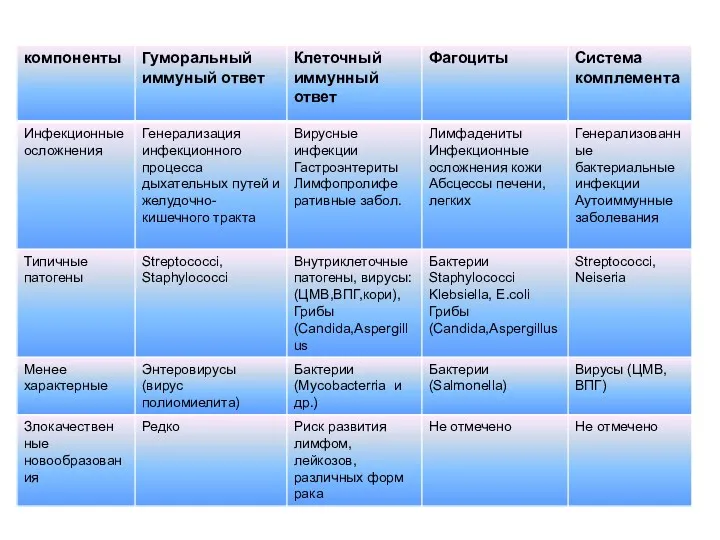
- 13. Типичные проявления ПИД Тяжело протекающие инфекции (75-100%) (бактериальные, вирусные, грибковые, вызванные необычными возбудителями; необычно протекающие); Аутоиммунные
- 14. Основные клинические характеристики ПИД Манифестация иммунодефицита с раннего возраста. Рецидивирующие инфекционные поражения ЛОР-органов и органов дыхания.
- 16. 10 настораживающих признаков ПИД: частый отит (6-8 раз в год); синусит (4-6 раз в год); пневмония
- 17. ОСНОВНЫЕ ПРИНЦИПЫ ДИАГНОСТИКИ ИД Сбор анамнеза Семейный анамнез. Осмотр (состояние лимфоидной ткани, размеры селезенки, печени и
- 18. I. ТКИН
- 19. I. Комбинированные Т- и В-клеточные ИД (ТКИН) Т–В+ТКИН (дефицит γ-цепи IL-2R; JAK3; CD45; α-цепи рецептора ИЛ-7;
- 20. I. ТКИН Клинические проявления: Наличие тяжелых, потенциально смертельных инфекций с 1-х недель жизни (Характерная триада: диарея,
- 21. А).Грибковые поражения кожи у больных ТКИН Развитие вакцинальной BCG-инфекции у пациентки с ТКИН
- 22. Лабораторные критерии: Лимфоцитопения (менее 1500 кл/мкл) Снижение или отсутствие Т и/или В-лимфоцитов в зависимости от формы
- 23. Х-сцепленная тяжелая комбинированная иммунная недостаточность (ТКИН) Частота: 1:100000 новорожденных (46% от всех ТКИН) Мутация гена γ-цепи
- 25. Тяжелая комбинированная иммунная недостаточность, обусловленная дефицитом JAK3 JAK3-киназа, сигнальная молекула, взаимодействующая с γ-цепью, дефект гена этой
- 26. Дефицит RAG1 и RAG2 . Мутации генов RAG1 и RAG2 блокируют процесс перестройки V-генов. Происходит блок
- 27. ТКИН ТКИН с дефицитом аденозиндезаминазы (1972г.) Мутация гена АДА (Известны более 50 мутаций). (АДА катализирует превращение
- 28. ТКИН ТКИН с дефицитом пуриннуклеозидфосфорилазы Аутосомно-рецессивный тип наследования, дефект гена пуриннуклеозидфосфорилазы. При дефиците ПНФ в клетках
- 29. Основные принципы терапии ТКИН После постановки диагноза дети должны находиться в стерильных боксах Интенсивная противомикробная, противовирусная
- 30. II. ИД с преимущественным нарушением продукции антител. XLA (болезнь Брутона) - (есть формы не связанные с
- 31. Заболевание проявляется на втором полугодии жизни ребенка Симптомы: возвратные инфекции верхних и нижних дыхательных путей, повторные
- 32. Динамика уровня различных классов иммуноглобулинов в детском возрасте.
- 33. II. ИД с преимущественным нарушением продукции антител. Х-сцепленная агаммаглобулинемия с дефицитом В-клеток Х-сцепленный вариант (85 %)
- 34. Нарушение дифференцировки В лимфоцитов при Х-сцепленной агаммаглобулинемии
- 35. Диагностика: Содержание CD19+ В-клеток в периферической крови Уменьшение содержания IgG в сыворотке крови (меньше 2 г/л)
- 36. II. Общая вариабельная иммунологическая недостаточность (ОВИН). ОВИН – сборная группа ПИД, и диагноз ставится после исключения
- 37. II. Общая вариабельная иммунологическая недостаточность (ОВИН). Клиническая картина: Часто диагностируется в возрасте 20-40 лет Рецидивирующие инфекции
- 38. Молекулярные дефекты при ОВИН Нарушения дифференцировки и созревания ДК Дефекты фагоцитоза (моноциты, макрофаги) Гиперпродукция ИЛ-12 Снижение
- 39. II. ИД с преимущественным нарушением продукции антител. Гипер-IgM-синдром (1974 г.) - редкая форма ПИД (Х-сцепленная и
- 40. Молекулярные механизмы развития Гипер-IgM синдрома Х-сцепленные формы Гипер IgM 1: мутация в гене CD40L, что приводит
- 41. III.Синдромы ИД с хорошо охарактеризованными клиническими признаками Дефекты репарации ДНК: Атаксия- телеангиэктазия (синдром Луи- Бар), синдром
- 42. III. Синдромы ИД с хорошо охарактеризованными клиническими признаками Атаксия-телеангиэктазия (синдром Луи-Бар) Аутосомно-рецессивное заболевание, частота: 1:500000 –
- 43. Атаксия-телеангиэктазия Клинические проявления: Прогрессирующая мозжечковая атаксия, неустойчивая походка, гипотония мышц; Телеангиэктазия мелких сосудов, расположенных в конъюктивах
- 44. Синдром Ниймеген Молекулярный дефект. Дефект гена NBS1(Nijmegen breakage syndrome), кодирующего синтез белка нибрина (хромосома 8q.21), участвующего
- 45. Синдром Вискотта-Олдрича (X-сцепленный ИД) Молекулярный дефект. Мутация гена WAS, отвечающего за выработку белка WASP (Wiskott-Aldrich Syndrome
- 46. Клинические проявления: Геморрагические проявления из-за тромбоцитопении: кровотечения, петехии; Экзема; Тяжелые инфекции (пневмоцистные пневмонии, герпетические инфекции), бактериальные
- 47. Молекулярный дефект Дефект гена Tbx1 в результате делеции в хромосоме 22. Ген Tbx1 кодирует транскрипционный фактор
- 48. Синдром Ди Джорджи Иммунологические нарушения: снижение числа СD3 клеток( В лимфоциты в норме; IgG,IgM – N;
- 49. IV . ИД с иммунной дисрегуляцией Х-сцепленный лимфопролиферативный синдром Аутоиммунный лимфопролиферативный синдром IPEX синдром APECED-синдром Синдром
- 50. IV . ИД с иммунной дисрегуляцией Аутоиммунный лимфопролиферативный синдром Дефект связан с нарушением апоптоза лимфоцитов. Симптоматика
- 51. Аутоиммунный лимфопролиферативный синдром Молекулярные дефекты: Дефект гена Fas (рецептора); Дефект гена FasL (лиганда); Дефект гена каспазы
- 52. Х-сцепленный лимфопролиферативный синдром. Редкий тяжелый ИД, характеризующийся неспособностью развивать иммунный ответ к EBV Молекулярный дефект. В
- 53. Клиническая картина. EBV вызывает поликлональную пролиферацию В,Т, моноцитов, эти клетки инфильтрируют печень,почки, вызывая нарушение функций этих
- 54. IPEX синдром (Immunodysregulation,Polyendocrinopathy and enteropathy, X-linked) Сцепленный с Х-хромосомой синдром дисрегуляции иммунитета, полиэндокринопатии и энтеропатии. Развивается
- 55. APECED синдром. Аутоиммунная полиэндокринопатия, кандидоз, эктодермальная дистрофия. Аутоиммунный синдром, обусловленный дефектом отрицательной селекции тимоцитов. Мутация гена
- 56. V. Врожденные дефекты фагоцитов (числа, функций или и того и другого) Тяжелая врожденная нейтропения, Циклическая нейтропения
- 57. Врожденные дефекты фагоцитов
- 58. Хроническая гранулематозная болезнь. Молекулярный дефект. Фагоциты больных не способны генерировать АФК, нарушается «кислородный взрыв», необходимый для
- 59. Хроническая гранулематозная болезнь Клинические проявления. Могут возникать у детей в раннем возрасте (1-1,5 года). Иногда у
- 60. Клинические проявления При хронической гранулематозной болезни у 15-30% больных развивается остеомиелит мелких костей пальцев рук и
- 61. Диагностика. Для иммунодиагностики ХГБ используют тесты, помогающие выявить нарушения фагоцитоза, (НСТ).В иммунограмме выявляют снижение высвобождения супероксида
- 62. VI. Дефекты врожденного иммунитета: рецепторов и сигнальных компонентов Нарушения TLR-сигнального пути (Дефект IRAK4-киназы, трансмембранного белка эндоплазматического
- 63. Дефекты врожденного иммунитета: рецепторы и сигнальные молекулы (0,4%) (2007)
- 64. СИГНАЛЬНЫЕ ПУТИ TLR IRAK1 TRAF6 IRAK4 CD 14 ЛПС TLR4 NEMO (IKKγ): полиморфизм чувствительности к вирусным
- 65. VII. Аутовоспалительные заболевания Причина воспаления - дисрегуляция врожденной иммунной системы из-за нарушений, в первую очередь, в
- 66. NOD1 рецептор экспрессируется практически во всех клетках организма, тогда как NOD2 рецептор экспрессируется преимущественно лейкоцитами периферической
- 67. ТРИ ОСНОВНЫХ ДОМЕНА: 1. РАСПОЗНАЮЩИЙ (LRR – ОБОГАЩЕН ЛЕЙЦИНОВЫМИ ПОВТОРАМИ). 2. ОЛИГОМЕРИЗУЮЩИЙ (NBD, NACHT, В НЕКОТОРЫХ
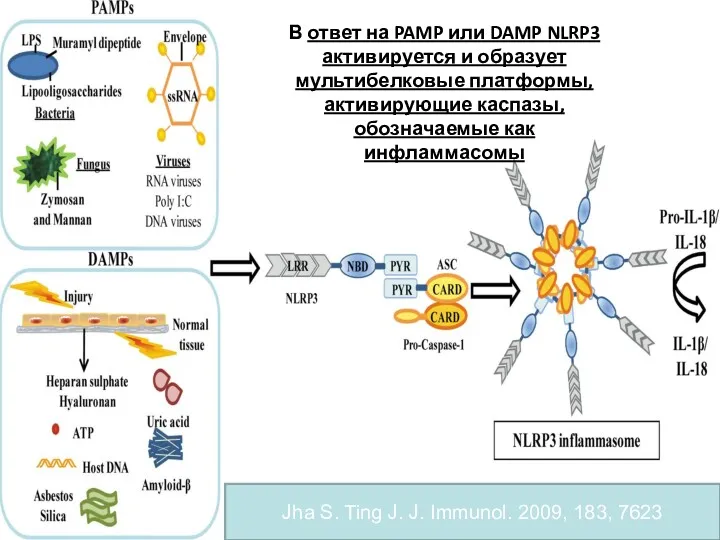
- 68. В ответ на распознавание PAMP и DAMP NLR белки могут участвовать в сборке мультибелкового комплекса, активирующего
- 69. ИНФЛАММАСОМЫ (INFLAMMASOMES) МОЛЕКУЛЯРНЫЕ СТРУКТУРЫ (ПЛАТФОРМЫ) ДЛЯ АКТИВЦИИ ВОСПАЛИТЕЛЬНЫХ КАСПАЗ И ДЛЯ ПРОЦЕССИНГА И СЕКРЕЦИИ ПРОВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ
- 70. Jha S. Ting J. J. Immunol. 2009, 183, 7623 В ответ на PAMP или DAMP NLRP3
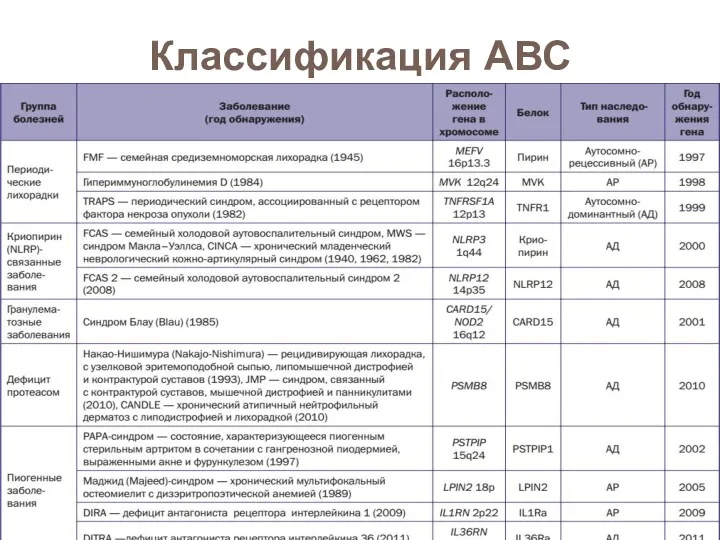
- 71. Классификация АВС
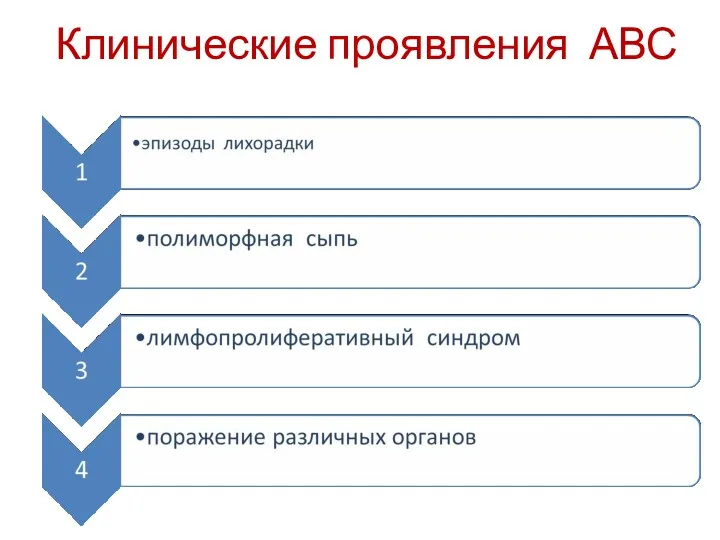
- 72. Клинические проявления АВС
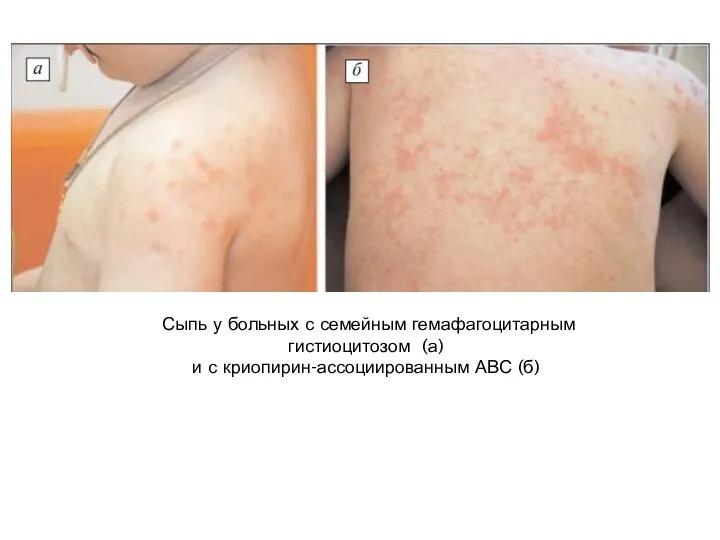
- 73. Сыпь у больных с семейным гемафагоцитарным гистиоцитозом (а) и с криопирин-ассоциированным АВС (б)
- 74. мутации в гене NLRP3, который кодирует белок криопирин аутосомно-доминантный тип наследования Криопиринассоциированный периодический синдром
- 75. СЕМЕЙНАЯ СРЕДИЗЕМНОМОРСКАЯ ЛИХОРАДКА (FAMILIAL MEDITERRANEAN FEVER, FMF) - один из наиболее исследованных синдромов, с которого началась
- 76. Появление генно-инженерных биологических препаратов значительно оптимизировало терапию FMF. Препараты, блокирующие ИЛ 1 (анакинра) и ФНО α
- 77. VIII. Дефекты в системе комплемента - Ангионевротический отек (дефицит С1-ингибитора) - Рецидивирующие пиогенные инфекции (дефицит С3,
- 78. Врожденные дефекты системы комплемента
- 80. TABLE I. Novel PID genes and their phenotypes (J Allergy Clin Immunol 2013;131:314-23.013) Gene Protein Inheritance
- 81. ОСНОВНЫЕ ПРИНЦИПЫ ЛЕЧЕНИЯ ПИД Трансплантация стволовых гемопоэтических и других клеток. 2. Заместительная терапия (Ig терапия, компоненты
- 82. Вторичные (приобретенные) иммунодефициты: нарушения иммунной системы, развившиеся в постнатальном периоде вследствие действия ненаследственных индукторных факторов (внутренних
- 83. Отличия первичных и вторичных иммунодефицитов
- 84. Механизмы развития ВИД: 1. гибель клеток иммунной системы по механизмам некроза и апоптоза. Причины некроза: неадекватные
- 85. Механизмы развития ВИД: 2. Нарушение функциональной активности лимфоцитов. 3. Дисбаланс регуляторных механизмов популяций. Может проявляться в
- 86. Клинические проявления ВИД: инфекционный, аллергический, аутоиммунный или иммунопролиферативный синдром Вторичные иммунодефициты могут развиться в любой период
- 87. Инфекции Нарушение питания (дефицит белка, витаминов, микроэлементов: цинка, селена и др.) Лекарственные вещества (цитостатики, иммунодепрессанты, антибиотики)
- 88. Классификация вторичных иммунодефицитов 1.Индуцированная форма Возникает в результате конкретных воздействий: рентгеновского излучения, цитостатической терапии, применения глюкокортикоидов,
- 89. Классификация вторичных иммунодефицитов 1.Острые Возникают при травмах, ожогах, стрессах, тяжелой острой вирусной или бактериальной инфекции и
- 90. Патогенные микроорганизмы, вызывающие развитие инфекционных заболеваний у лиц, получающих иммуносупрессивную терапию.
- 91. Физиологические иммунодефициты
- 92. При стрессе: В основе стресса лежит повышенная выработка АКТГ, выброс глюкокортикоидов и катехоламинов. При умеренном воздействии
- 93. При интенсивных стрессорных воздействиях выброс гормонов переходит границу, при которой еще не происходит апоптоза. Устойчивость клеток
- 94. Также происходит подавление функции макрофагов, что частично обусловлено увеличением внутриклеточной концентрации цАМФ. Результатом всех этих изменений
- 95. Возрастные иммунодефициты: 1. Иммунодефицит раннего постнатального периода. Связан с тем, что формирование иммунной системы не завершается
- 96. Недостаточность ИФНγ влияет на функцию макрофагов, а низкая секреторная активность Th2 и недостаточность косигналов – на
- 97. Собственный синтез IgG появляется к 6 месяцам, полного развития достигает к 5 – 6 годам. Способность
- 98. Возрастные иммунодефициты: 2. Старение иммунной системы и связанный с ним иммунодефицит. Изменения в иммунной системе, приводящие
- 99. Основные проявление возрастных изменений тимусзависимой системы: а) передача функции тимуса на периферию. Т.е. повышается роль периферии
- 100. б) Снижение способности тимуса привлекать клетки – предшественники и его «пропускающей способности» в отношении созревающих клеток.
- 101. д) Функциональная недостаточность периферических Т-кл., из-за дефицита гормонов тимуса. е) снижение численности Т-клеток на периферии регистрируется
- 102. Ослабление иммунной защиты в основном затрагивает реакции, обусловленные Т-клетками, хотя явного роста заболеваемости не происходит. Увеличивается
- 103. Процесс старения иммунной системы может быть ускорен неблагоприятными факторами среда. Иммунологические изменения однонаправлены и необратимы. Среди
- 105. Скачать презентацию
ИММУНОДЕФИЦИТНЫЕ
ЗАБОЛЕВАНИЯ
ПЕРВИЧНЫЕ ВТОРИЧНЫЕ
(Врожденные), (Приобретенные),
основанные на формирующиеся под
генетических
ЗАБОЛЕВАНИЯ
ПЕРВИЧНЫЕ ВТОРИЧНЫЕ
(Врожденные), (Приобретенные),
основанные на формирующиеся под
генетических

ПЕРВИЧНЫЕ ИММУНОДЕФИЦИТЫ -
Заболевания иммунной системы, которые развиваются
в результате генетически
ПЕРВИЧНЫЕ ИММУНОДЕФИЦИТЫ -
Заболевания иммунной системы, которые развиваются
в результате генетически

Первый случай иммунодефицита описал военный педиатр армии США
полковник Ogden Carr
Первый случай иммунодефицита описал военный педиатр армии США
полковник Ogden Carr

«ИММУНОДЕФИЦИТЫ НАШИ УЧИТЕЛЯ»
«ИММУНОДЕФИЦИТЫ ПРОДОЛЖАЮТ
НАС УЧИТЬ»
Роберт Алан ГУД (Good)
(1922-2003)
АМЕРИКАНСКИЙ ПЕДИАТР - ОДИН
«ИММУНОДЕФИЦИТЫ ПРОДОЛЖАЮТ
НАС УЧИТЬ»
Роберт Алан ГУД (Good)
(1922-2003)
АМЕРИКАНСКИЙ ПЕДИАТР - ОДИН

Частота встречаемости ПИД соответствует другим генетическим дефектам человека (1:10000-15000)
селективный
Частота встречаемости ПИД соответствует другим генетическим дефектам человека (1:10000-15000)
селективный

Основные этапы изучения первичных иммунодефицитов
Изучение ПИД началось в 50-х годах, когда
Основные этапы изучения первичных иммунодефицитов
Изучение ПИД началось в 50-х годах, когда

КЛАССИФИКАЦИИ ПЕРВИЧНЫХ
ИММУНОДЕФИЦИТОВ (по данным ВОЗ)
1. 1968 – поражение клеточного и гуморального
КЛАССИФИКАЦИИ ПЕРВИЧНЫХ
ИММУНОДЕФИЦИТОВ (по данным ВОЗ)
1. 1968 – поражение клеточного и гуморального

ОТЕЧЕСТВЕННАЯ КЛАССИФИКАЦИЯ
ПЕРВИЧНЫХ ИММУНОДЕФИЦИТОВ
ОСНОВАНА НА ГЕНЕТИЧЕСКИХ БЛОКАХ
РАЗВИТИЯ Т- И В-КЛЕТОК
(по крайней
ОТЕЧЕСТВЕННАЯ КЛАССИФИКАЦИЯ
ПЕРВИЧНЫХ ИММУНОДЕФИЦИТОВ
ОСНОВАНА НА ГЕНЕТИЧЕСКИХ БЛОКАХ
РАЗВИТИЯ Т- И В-КЛЕТОК
(по крайней

Классификация первичных ИД (2007г.)
1. Комбинированные Т- и В-клеточные
иммунодефициты (8,4%);
2. ИД
Классификация первичных ИД (2007г.)
1. Комбинированные Т- и В-клеточные
иммунодефициты (8,4%);
2. ИД

9. Фенокопии ПИД
Наследуемые иммунодефициты, не связанные с мутациями в генах зародышевой
9. Фенокопии ПИД
Наследуемые иммунодефициты, не связанные с мутациями в генах зародышевой

Типичные проявления ПИД
Тяжело протекающие инфекции (75-100%) (бактериальные, вирусные, грибковые, вызванные
Типичные проявления ПИД
Тяжело протекающие инфекции (75-100%) (бактериальные, вирусные, грибковые, вызванные

Основные клинические характеристики ПИД
Манифестация иммунодефицита с раннего возраста.
Рецидивирующие инфекционные
Основные клинические характеристики ПИД
Манифестация иммунодефицита с раннего возраста.
Рецидивирующие инфекционные

10 настораживающих признаков ПИД:
частый отит (6-8 раз в год);
синусит (4-6
10 настораживающих признаков ПИД:
частый отит (6-8 раз в год);
синусит (4-6

ОСНОВНЫЕ ПРИНЦИПЫ ДИАГНОСТИКИ ИД
Сбор анамнеза
Семейный анамнез.
Осмотр (состояние лимфоидной ткани,
ОСНОВНЫЕ ПРИНЦИПЫ ДИАГНОСТИКИ ИД
Сбор анамнеза
Семейный анамнез.
Осмотр (состояние лимфоидной ткани,
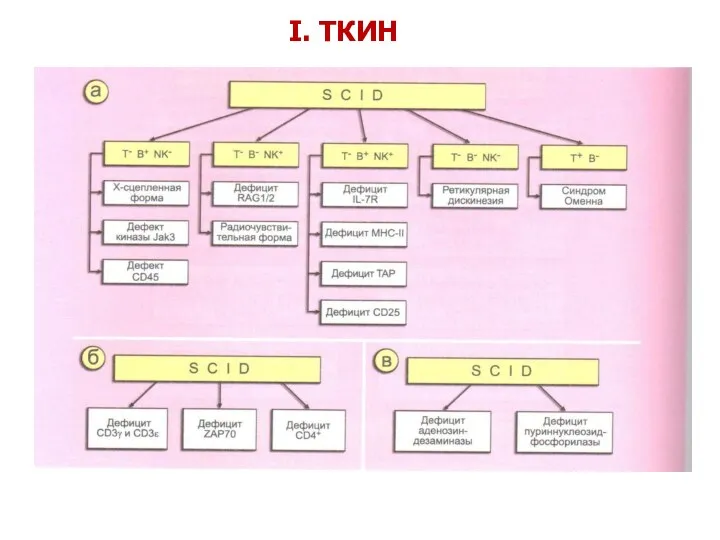
I. ТКИН
I. ТКИН

I. Комбинированные Т- и В-клеточные ИД
(ТКИН)
Т–В+ТКИН
(дефицит γ-цепи IL-2R; JAK3;
I. Комбинированные Т- и В-клеточные ИД
(ТКИН)
Т–В+ТКИН
(дефицит γ-цепи IL-2R; JAK3;

I. ТКИН
Клинические проявления:
Наличие тяжелых, потенциально смертельных инфекций с 1-х недель жизни
I. ТКИН
Клинические проявления:
Наличие тяжелых, потенциально смертельных инфекций с 1-х недель жизни
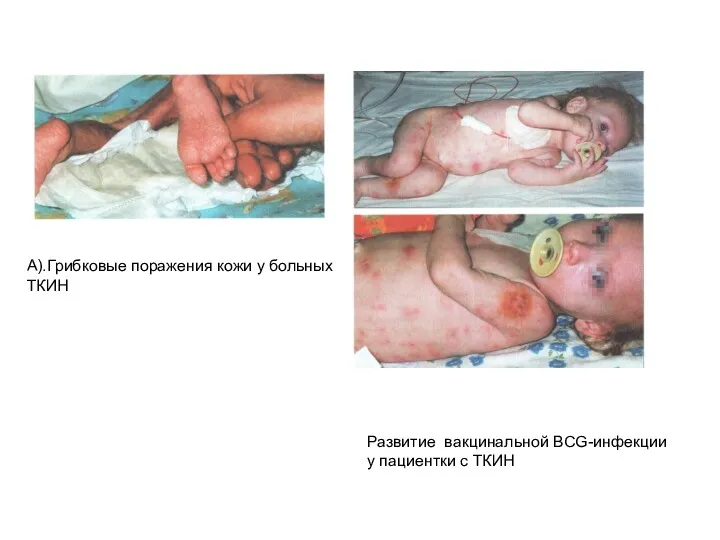
А).Грибковые поражения кожи у больных
ТКИН
Развитие вакцинальной BCG-инфекции
у пациентки с ТКИН
А).Грибковые поражения кожи у больных
ТКИН
Развитие вакцинальной BCG-инфекции
у пациентки с ТКИН

Лабораторные критерии:
Лимфоцитопения (менее 1500 кл/мкл)
Снижение или отсутствие Т и/или В-лимфоцитов
Лабораторные критерии:
Лимфоцитопения (менее 1500 кл/мкл)
Снижение или отсутствие Т и/или В-лимфоцитов
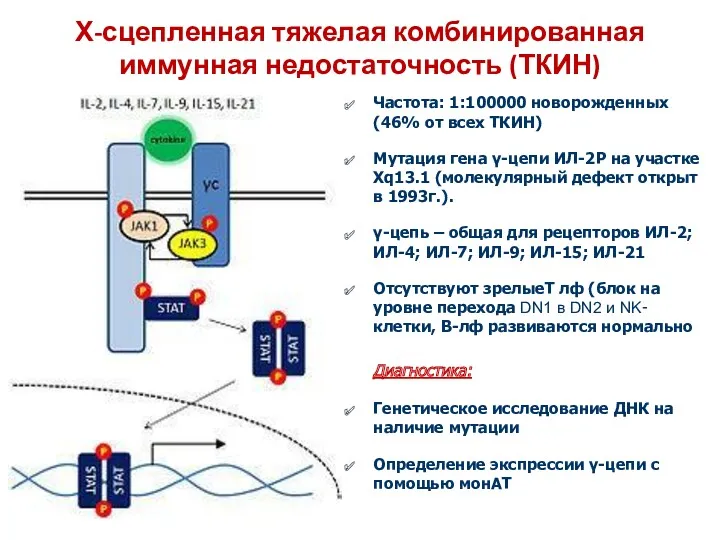
Х-сцепленная тяжелая комбинированная иммунная недостаточность (ТКИН)
Частота: 1:100000 новорожденных (46% от всех
Х-сцепленная тяжелая комбинированная иммунная недостаточность (ТКИН)
Частота: 1:100000 новорожденных (46% от всех

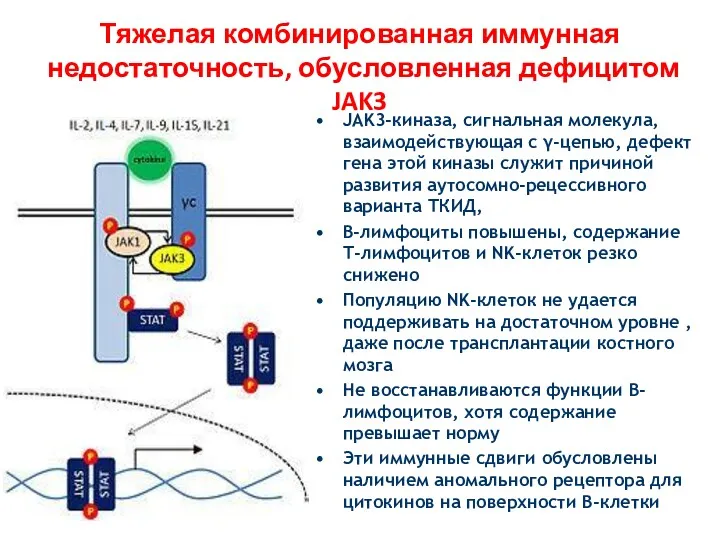
Тяжелая комбинированная иммунная недостаточность, обусловленная дефицитом JAK3
JAK3-киназа, сигнальная молекула, взаимодействующая с
Тяжелая комбинированная иммунная недостаточность, обусловленная дефицитом JAK3
JAK3-киназа, сигнальная молекула, взаимодействующая с

Дефицит RAG1 и RAG2 .
Мутации генов RAG1 и RAG2
Дефицит RAG1 и RAG2 .
Мутации генов RAG1 и RAG2

ТКИН
ТКИН с дефицитом аденозиндезаминазы (1972г.)
Мутация гена АДА (Известны более
ТКИН
ТКИН с дефицитом аденозиндезаминазы (1972г.)
Мутация гена АДА (Известны более

ТКИН
ТКИН с дефицитом пуриннуклеозидфосфорилазы
Аутосомно-рецессивный тип наследования, дефект гена пуриннуклеозидфосфорилазы. При дефиците
ТКИН
ТКИН с дефицитом пуриннуклеозидфосфорилазы
Аутосомно-рецессивный тип наследования, дефект гена пуриннуклеозидфосфорилазы. При дефиците

Основные принципы терапии ТКИН
После постановки диагноза дети
должны находиться в стерильных
Основные принципы терапии ТКИН
После постановки диагноза дети
должны находиться в стерильных

II. ИД с преимущественным нарушением
продукции антител.
XLA (болезнь Брутона)
продукции антител.
XLA (болезнь Брутона)

Заболевание проявляется на втором полугодии жизни ребенка
Симптомы:
возвратные инфекции
Заболевание проявляется на втором полугодии жизни ребенка
Симптомы:
возвратные инфекции
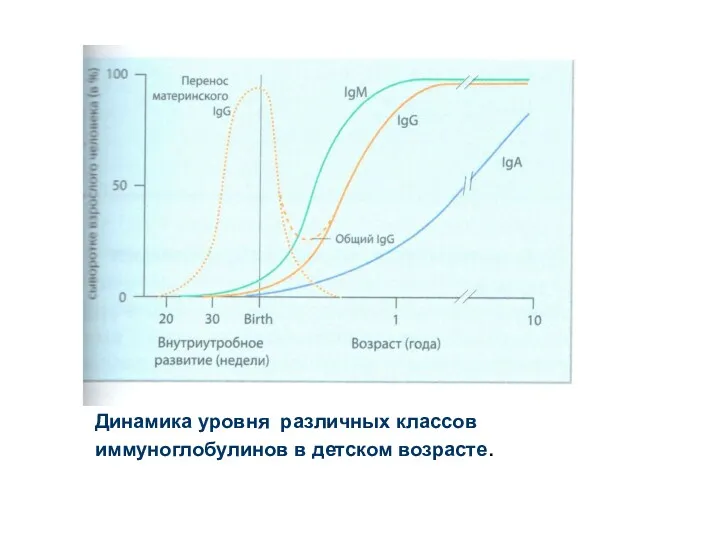
Динамика уровня различных классов
иммуноглобулинов в детском возрасте.
Динамика уровня различных классов
иммуноглобулинов в детском возрасте.

II. ИД с преимущественным нарушением продукции антител.
Х-сцепленная агаммаглобулинемия с дефицитом
II. ИД с преимущественным нарушением продукции антител.
Х-сцепленная агаммаглобулинемия с дефицитом
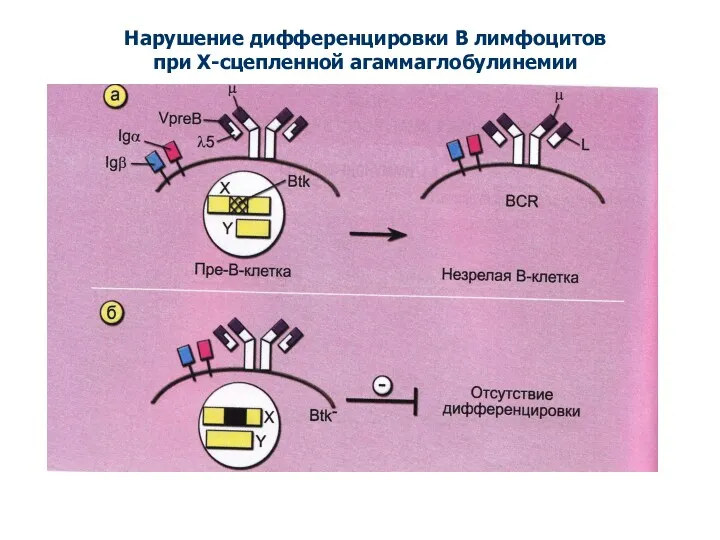
Нарушение дифференцировки В лимфоцитов
при Х-сцепленной агаммаглобулинемии
Нарушение дифференцировки В лимфоцитов
при Х-сцепленной агаммаглобулинемии

Диагностика:
Содержание CD19+ В-клеток в периферической крови < 1-2 %
Уменьшение содержания IgG
Диагностика:
Содержание CD19+ В-клеток в периферической крови < 1-2 %
Уменьшение содержания IgG

II. Общая вариабельная иммунологическая недостаточность (ОВИН).
ОВИН – сборная группа ПИД, и
II. Общая вариабельная иммунологическая недостаточность (ОВИН).
ОВИН – сборная группа ПИД, и

II. Общая вариабельная иммунологическая недостаточность (ОВИН).
Клиническая картина:
Часто диагностируется в возрасте 20-40
II. Общая вариабельная иммунологическая недостаточность (ОВИН).
Клиническая картина:
Часто диагностируется в возрасте 20-40
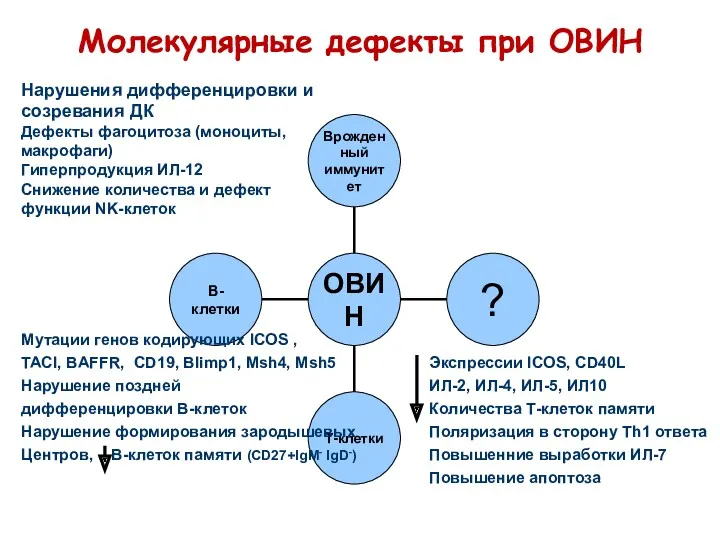
Молекулярные дефекты при ОВИН
Нарушения дифференцировки и
созревания ДК
Дефекты фагоцитоза (моноциты,
макрофаги)
Гиперпродукция ИЛ-12
Снижение количества
Молекулярные дефекты при ОВИН
Нарушения дифференцировки и
созревания ДК
Дефекты фагоцитоза (моноциты,
макрофаги)
Гиперпродукция ИЛ-12
Снижение количества

II. ИД с преимущественным нарушением продукции антител.
Гипер-IgM-синдром (1974 г.)
II. ИД с преимущественным нарушением продукции антител.
Гипер-IgM-синдром (1974 г.)
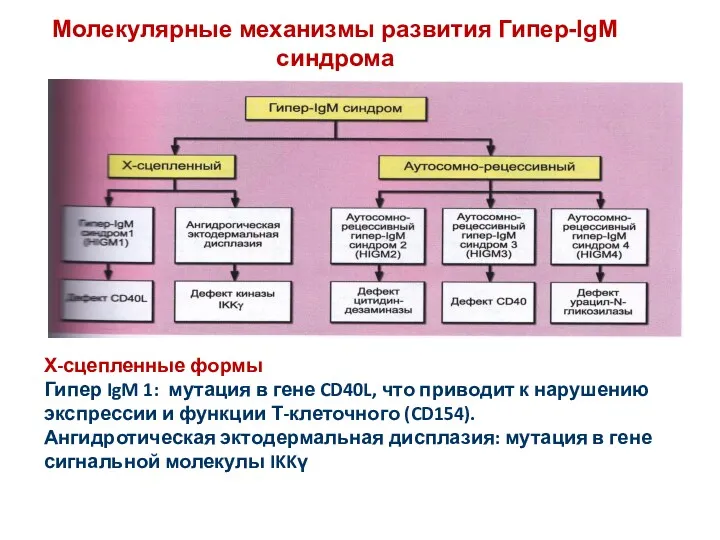
Молекулярные механизмы развития Гипер-IgM синдрома
Х-сцепленные формы
Гипер IgM 1: мутация в
Молекулярные механизмы развития Гипер-IgM синдрома
Х-сцепленные формы
Гипер IgM 1: мутация в

III.Синдромы ИД с хорошо охарактеризованными клиническими признаками
Дефекты репарации ДНК:
Дефекты репарации ДНК:

III. Синдромы ИД с хорошо охарактеризованными клиническими признаками
Атаксия-телеангиэктазия (синдром
III. Синдромы ИД с хорошо охарактеризованными клиническими признаками
Атаксия-телеангиэктазия (синдром
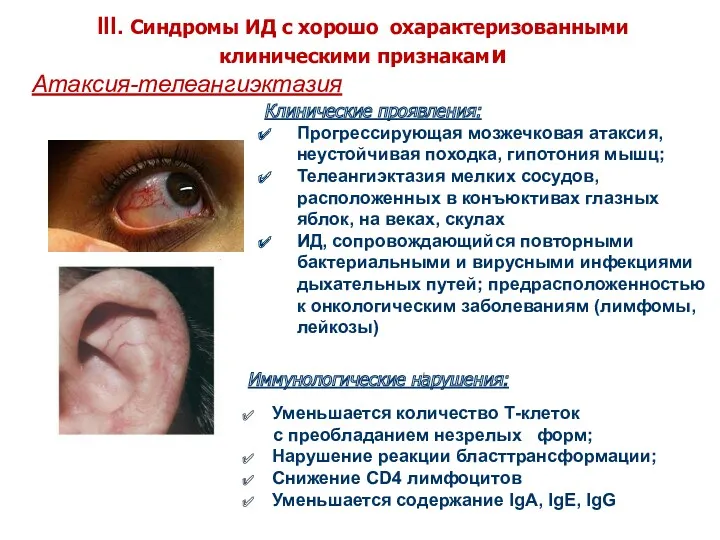
Атаксия-телеангиэктазия
Клинические проявления:
Прогрессирующая мозжечковая атаксия, неустойчивая походка, гипотония мышц;
Телеангиэктазия мелких сосудов,
Атаксия-телеангиэктазия
Клинические проявления:
Прогрессирующая мозжечковая атаксия, неустойчивая походка, гипотония мышц;
Телеангиэктазия мелких сосудов,
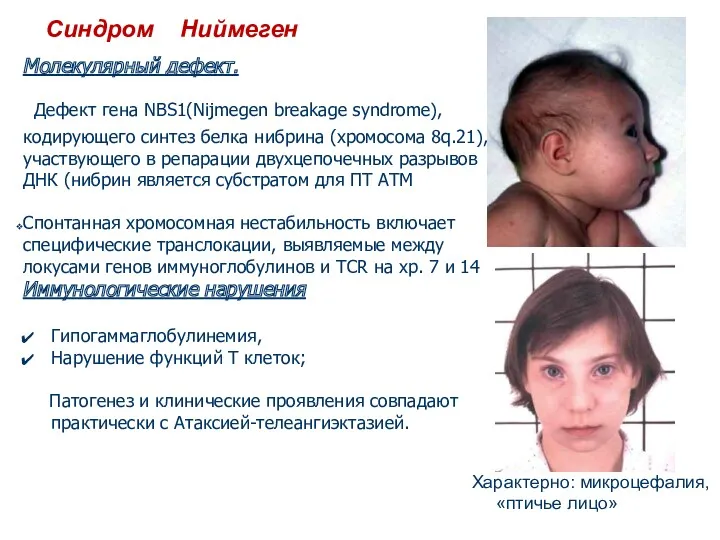
Синдром Ниймеген
Молекулярный дефект.
Дефект гена NBS1(Nijmegen breakage syndrome), кодирующего синтез
Синдром Ниймеген
Молекулярный дефект.
Дефект гена NBS1(Nijmegen breakage syndrome), кодирующего синтез
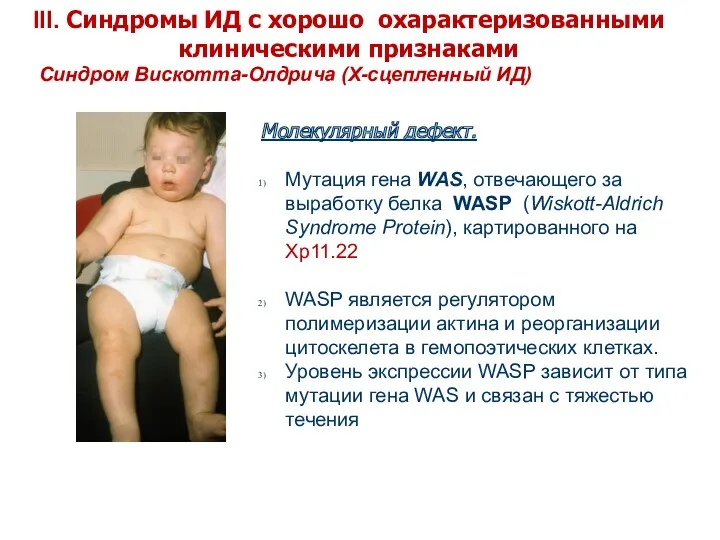
Синдром Вискотта-Олдрича (X-сцепленный ИД)
Молекулярный дефект.
Мутация гена WAS, отвечающего за выработку
Молекулярный дефект.
Мутация гена WAS, отвечающего за выработку
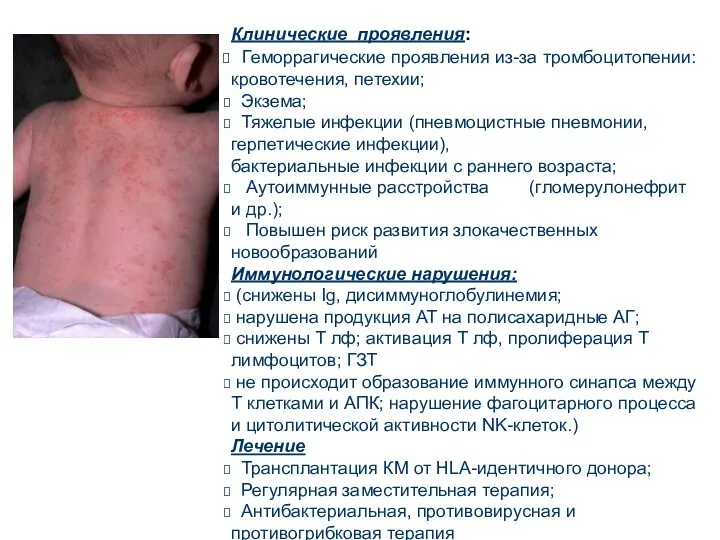
Клинические проявления:
Геморрагические проявления из-за тромбоцитопении: кровотечения, петехии;
Экзема;
Тяжелые
Клинические проявления:
Геморрагические проявления из-за тромбоцитопении: кровотечения, петехии;
Экзема;
Тяжелые
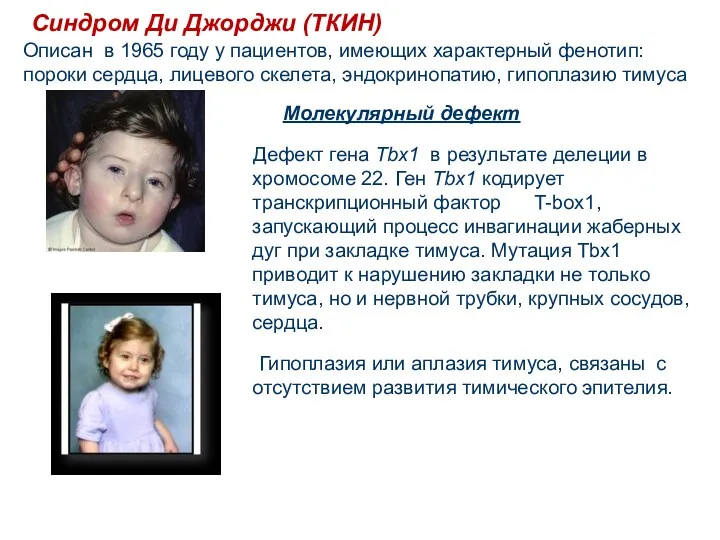
Молекулярный дефект
Дефект гена Tbx1 в результате делеции в хромосоме
Молекулярный дефект
Дефект гена Tbx1 в результате делеции в хромосоме

Синдром Ди Джорджи
Иммунологические нарушения:
снижение числа СD3 клеток(< 500/мкл);CD4,CD8;
В лимфоциты в норме;
Синдром Ди Джорджи
Иммунологические нарушения:
снижение числа СD3 клеток(< 500/мкл);CD4,CD8;
В лимфоциты в норме;

IV . ИД с иммунной дисрегуляцией
Х-сцепленный лимфопролиферативный синдром
Аутоиммунный
IV . ИД с иммунной дисрегуляцией
Х-сцепленный лимфопролиферативный синдром
Аутоиммунный

IV . ИД с иммунной дисрегуляцией
Аутоиммунный лимфопролиферативный синдром
Дефект связан
IV . ИД с иммунной дисрегуляцией Аутоиммунный лимфопролиферативный синдром Дефект связан

Аутоиммунный лимфопролиферативный синдром
Молекулярные дефекты:
Дефект гена Fas (рецептора);
Дефект гена FasL
Аутоиммунный лимфопролиферативный синдром
Молекулярные дефекты:
Дефект гена Fas (рецептора);
Дефект гена FasL

Х-сцепленный лимфопролиферативный синдром.
Редкий тяжелый ИД, характеризующийся неспособностью развивать иммунный
Х-сцепленный лимфопролиферативный синдром. Редкий тяжелый ИД, характеризующийся неспособностью развивать иммунный

Клиническая картина.
EBV вызывает поликлональную пролиферацию В,Т, моноцитов, эти клетки инфильтрируют
Клиническая картина.
EBV вызывает поликлональную пролиферацию В,Т, моноцитов, эти клетки инфильтрируют

IPEX синдром (Immunodysregulation,Polyendocrinopathy and enteropathy, X-linked)
Сцепленный с Х-хромосомой синдром дисрегуляции
IPEX синдром (Immunodysregulation,Polyendocrinopathy and enteropathy, X-linked)
Сцепленный с Х-хромосомой синдром дисрегуляции

APECED синдром.
Аутоиммунная полиэндокринопатия, кандидоз, эктодермальная дистрофия. Аутоиммунный синдром, обусловленный
APECED синдром.
Аутоиммунная полиэндокринопатия, кандидоз, эктодермальная дистрофия. Аутоиммунный синдром, обусловленный

V. Врожденные дефекты фагоцитов
(числа, функций или и того и другого)
V. Врожденные дефекты фагоцитов
(числа, функций или и того и другого)
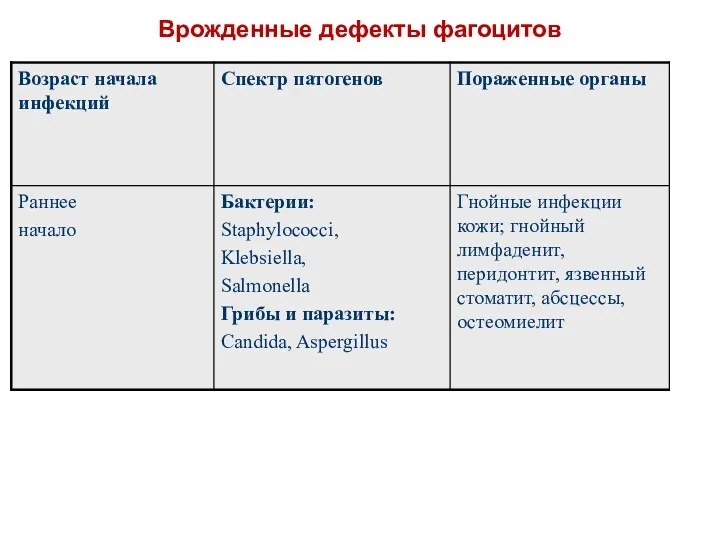
Врожденные дефекты фагоцитов
Врожденные дефекты фагоцитов

Хроническая гранулематозная болезнь.
Молекулярный дефект.
Фагоциты больных не способны генерировать АФК, нарушается
Хроническая гранулематозная болезнь.
Молекулярный дефект.
Фагоциты больных не способны генерировать АФК, нарушается

Хроническая гранулематозная болезнь
Клинические проявления.
Могут возникать у детей в раннем
Хроническая гранулематозная болезнь
Клинические проявления.
Могут возникать у детей в раннем

Клинические проявления
При хронической гранулематозной болезни у 15-30% больных развивается
Клинические проявления
При хронической гранулематозной болезни у 15-30% больных развивается

Диагностика. Для иммунодиагностики ХГБ используют тесты, помогающие выявить нарушения фагоцитоза, (НСТ).В
Диагностика. Для иммунодиагностики ХГБ используют тесты, помогающие выявить нарушения фагоцитоза, (НСТ).В

VI. Дефекты врожденного иммунитета: рецепторов и сигнальных компонентов
Нарушения TLR-сигнального пути
(Дефект
VI. Дефекты врожденного иммунитета: рецепторов и сигнальных компонентов
Нарушения TLR-сигнального пути
(Дефект
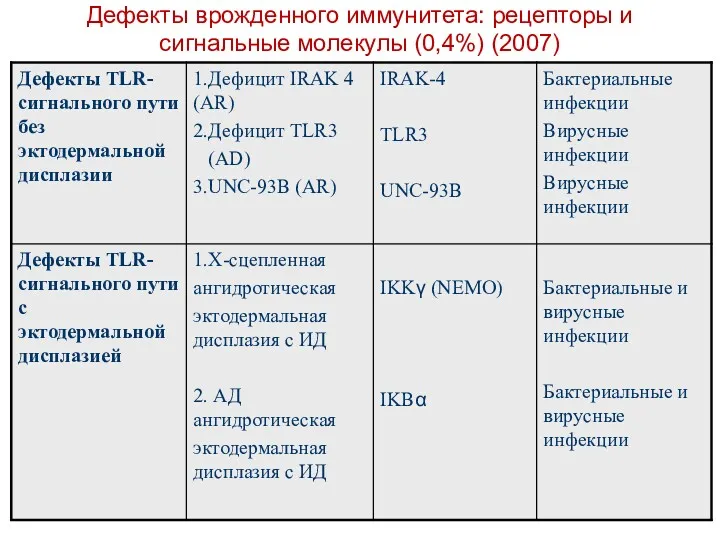
Дефекты врожденного иммунитета: рецепторы и сигнальные молекулы (0,4%) (2007)
Дефекты врожденного иммунитета: рецепторы и сигнальные молекулы (0,4%) (2007)
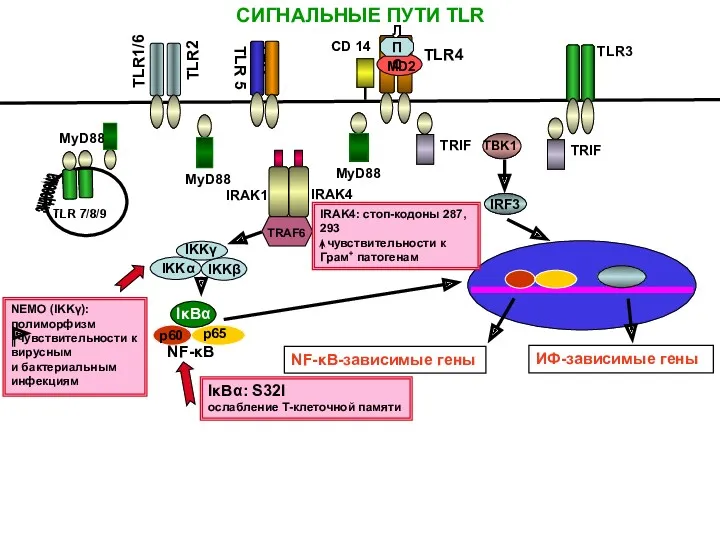
СИГНАЛЬНЫЕ ПУТИ TLR
IRAK1
TRAF6
IRAK4
CD 14
ЛПС
TLR4
NEMO (IKKγ): полиморфизм
чувствительности к вирусным
и бактериальным
СИГНАЛЬНЫЕ ПУТИ TLR
IRAK1
TRAF6
IRAK4
CD 14
ЛПС
TLR4
NEMO (IKKγ): полиморфизм
чувствительности к вирусным
и бактериальным

VII. Аутовоспалительные заболевания
Причина воспаления - дисрегуляция врожденной иммунной системы из-за нарушений,
VII. Аутовоспалительные заболевания
Причина воспаления - дисрегуляция врожденной иммунной системы из-за нарушений,

NOD1 рецептор экспрессируется практически во всех клетках организма, тогда как
NOD2
NOD1 рецептор экспрессируется практически во всех клетках организма, тогда как
NOD2
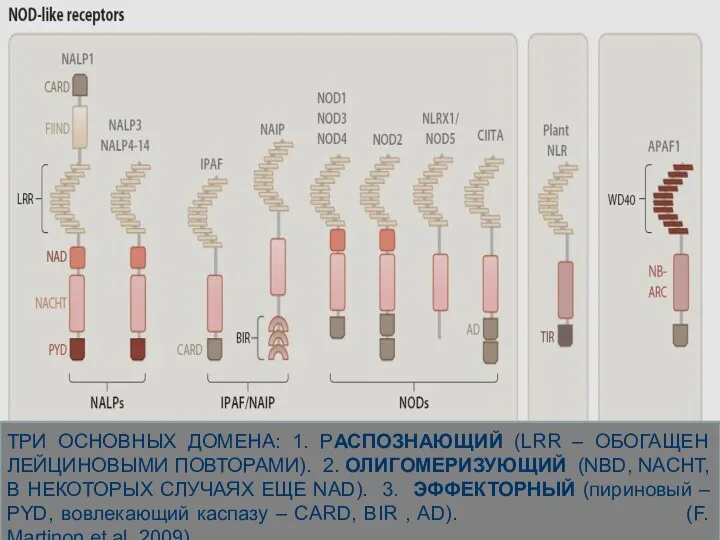
ТРИ ОСНОВНЫХ ДОМЕНА: 1. РАСПОЗНАЮЩИЙ (LRR – ОБОГАЩЕН ЛЕЙЦИНОВЫМИ ПОВТОРАМИ). 2.
ТРИ ОСНОВНЫХ ДОМЕНА: 1. РАСПОЗНАЮЩИЙ (LRR – ОБОГАЩЕН ЛЕЙЦИНОВЫМИ ПОВТОРАМИ). 2.

В ответ на распознавание
PAMP и DAMP
NLR белки могут участвовать
В ответ на распознавание
PAMP и DAMP
NLR белки могут участвовать

ИНФЛАММАСОМЫ (INFLAMMASOMES)
МОЛЕКУЛЯРНЫЕ СТРУКТУРЫ (ПЛАТФОРМЫ)
ДЛЯ АКТИВЦИИ ВОСПАЛИТЕЛЬНЫХ КАСПАЗ И
ДЛЯ ПРОЦЕССИНГА
ИНФЛАММАСОМЫ (INFLAMMASOMES)
МОЛЕКУЛЯРНЫЕ СТРУКТУРЫ (ПЛАТФОРМЫ)
ДЛЯ АКТИВЦИИ ВОСПАЛИТЕЛЬНЫХ КАСПАЗ И
ДЛЯ ПРОЦЕССИНГА
Jha S. Ting J. J. Immunol. 2009, 183, 7623
В ответ на
Jha S. Ting J. J. Immunol. 2009, 183, 7623
В ответ на
Классификация АВС
Классификация АВС
Клинические проявления АВС
Клинические проявления АВС
Сыпь у больных с семейным гемафагоцитарным гистиоцитозом (а)
и с криопирин-ассоциированным
Сыпь у больных с семейным гемафагоцитарным гистиоцитозом (а)
и с криопирин-ассоциированным
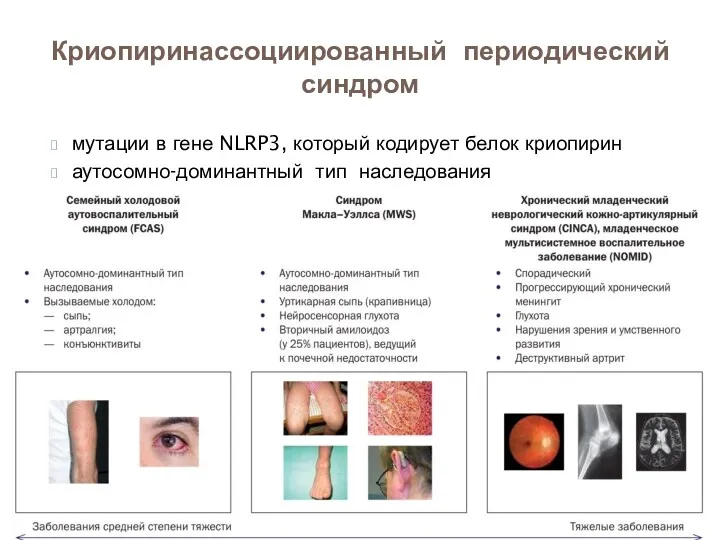
мутации в гене NLRP3, который кодирует белок криопирин
аутосомно-доминантный тип наследования
Криопиринассоциированный
мутации в гене NLRP3, который кодирует белок криопирин
аутосомно-доминантный тип наследования
Криопиринассоциированный

СЕМЕЙНАЯ СРЕДИЗЕМНОМОРСКАЯ ЛИХОРАДКА
(FAMILIAL MEDITERRANEAN FEVER, FMF) - один из наиболее
СЕМЕЙНАЯ СРЕДИЗЕМНОМОРСКАЯ ЛИХОРАДКА (FAMILIAL MEDITERRANEAN FEVER, FMF) - один из наиболее
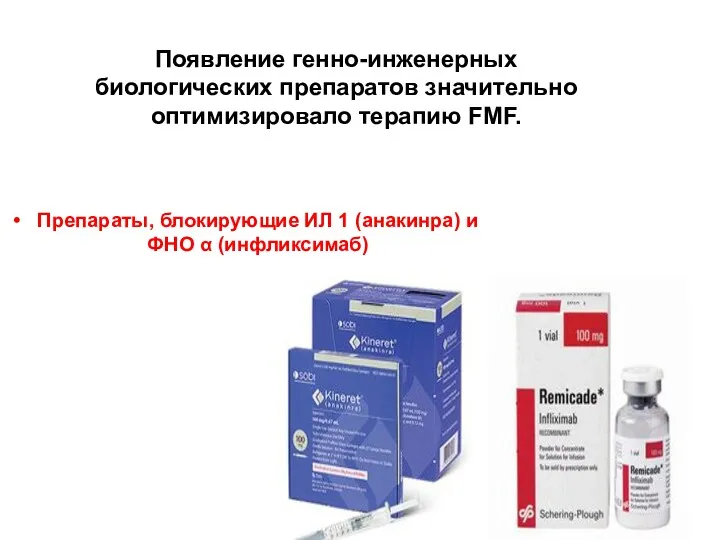
Появление генно-инженерных биологических препаратов значительно оптимизировало терапию FMF.
Препараты, блокирующие ИЛ
Появление генно-инженерных биологических препаратов значительно оптимизировало терапию FMF.
Препараты, блокирующие ИЛ

VIII. Дефекты в системе комплемента
- Ангионевротический отек (дефицит С1-ингибитора)
- Рецидивирующие
VIII. Дефекты в системе комплемента
- Ангионевротический отек (дефицит С1-ингибитора)
- Рецидивирующие
Врожденные дефекты системы комплемента
Врожденные дефекты системы комплемента
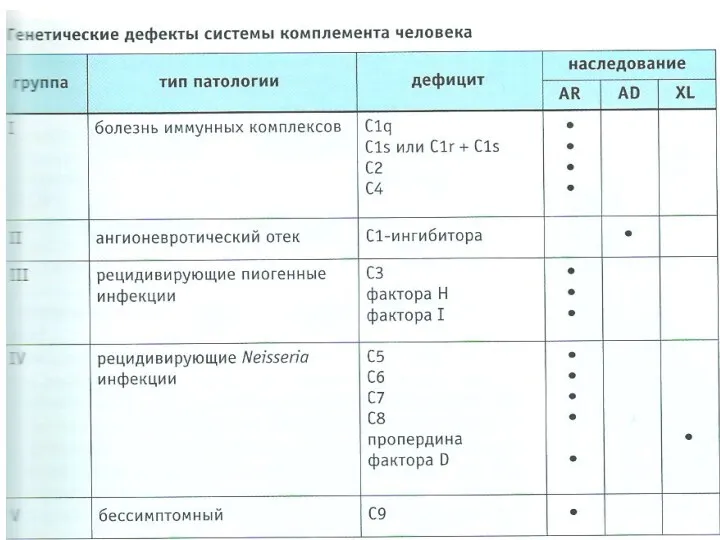
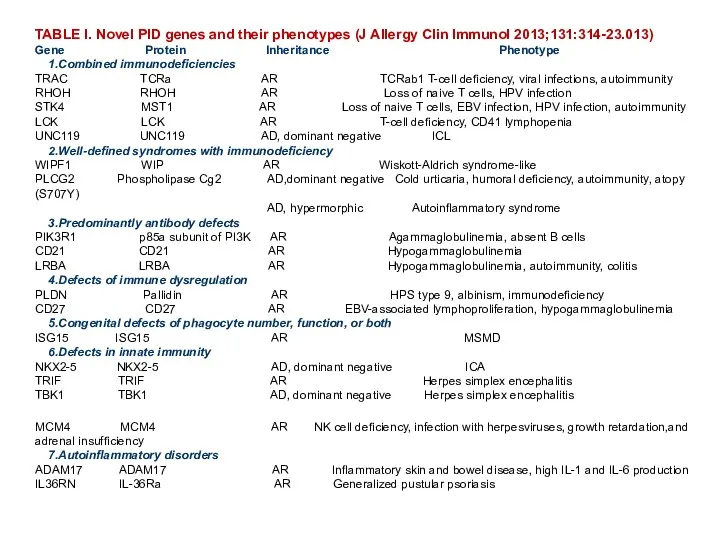
TABLE I. Novel PID genes and their phenotypes (J Allergy Clin
TABLE I. Novel PID genes and their phenotypes (J Allergy Clin

ОСНОВНЫЕ ПРИНЦИПЫ ЛЕЧЕНИЯ ПИД
Трансплантация стволовых гемопоэтических
и других клеток.
2. Заместительная терапия (Ig
ОСНОВНЫЕ ПРИНЦИПЫ ЛЕЧЕНИЯ ПИД
Трансплантация стволовых гемопоэтических
и других клеток.
2. Заместительная терапия (Ig

Вторичные (приобретенные) иммунодефициты:
нарушения иммунной системы, развившиеся в постнатальном периоде вследствие
Вторичные (приобретенные) иммунодефициты:
нарушения иммунной системы, развившиеся в постнатальном периоде вследствие

Отличия первичных и вторичных иммунодефицитов
Отличия первичных и вторичных иммунодефицитов

Механизмы развития ВИД:
1. гибель клеток иммунной системы по механизмам некроза и
Механизмы развития ВИД:
1. гибель клеток иммунной системы по механизмам некроза и

Механизмы развития ВИД:
2. Нарушение функциональной активности лимфоцитов.
3. Дисбаланс регуляторных механизмов популяций.
Может
Механизмы развития ВИД:
2. Нарушение функциональной активности лимфоцитов.
3. Дисбаланс регуляторных механизмов популяций.
Может

Клинические проявления ВИД: инфекционный, аллергический, аутоиммунный или иммунопролиферативный синдром
Вторичные
Клинические проявления ВИД: инфекционный, аллергический, аутоиммунный или иммунопролиферативный синдром
Вторичные

Инфекции
Нарушение питания (дефицит белка, витаминов, микроэлементов: цинка, селена и др.)
Лекарственные вещества
Инфекции
Нарушение питания (дефицит белка, витаминов, микроэлементов: цинка, селена и др.)
Лекарственные вещества

Классификация вторичных иммунодефицитов
1.Индуцированная форма
Возникает в результате конкретных воздействий: рентгеновского излучения,
Классификация вторичных иммунодефицитов
1.Индуцированная форма
Возникает в результате конкретных воздействий: рентгеновского излучения,

Классификация вторичных иммунодефицитов
1.Острые
Возникают при травмах, ожогах, стрессах, тяжелой острой вирусной
Классификация вторичных иммунодефицитов
1.Острые
Возникают при травмах, ожогах, стрессах, тяжелой острой вирусной
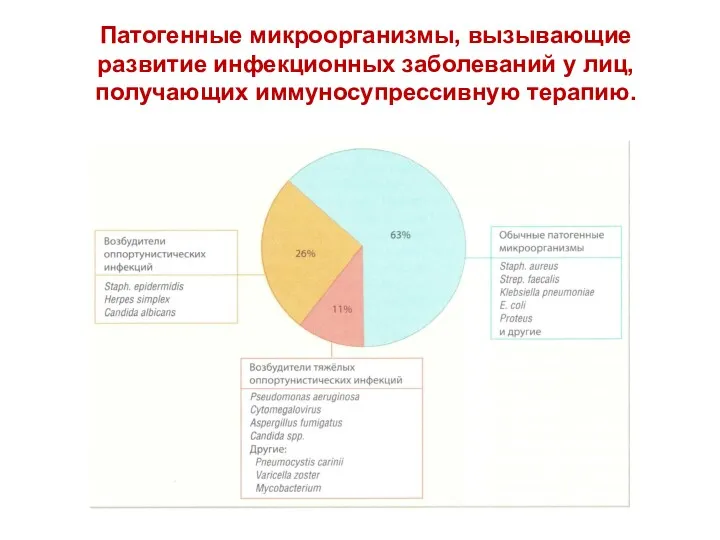
Патогенные микроорганизмы, вызывающие развитие инфекционных заболеваний у лиц, получающих иммуносупрессивную терапию.
Патогенные микроорганизмы, вызывающие развитие инфекционных заболеваний у лиц, получающих иммуносупрессивную терапию.

Физиологические иммунодефициты
Физиологические иммунодефициты

При стрессе:
В основе стресса лежит повышенная выработка АКТГ, выброс глюкокортикоидов и
При стрессе:
В основе стресса лежит повышенная выработка АКТГ, выброс глюкокортикоидов и

При интенсивных стрессорных воздействиях выброс гормонов переходит границу, при которой еще
При интенсивных стрессорных воздействиях выброс гормонов переходит границу, при которой еще

Также происходит подавление функции макрофагов, что частично обусловлено увеличением внутриклеточной концентрации
Также происходит подавление функции макрофагов, что частично обусловлено увеличением внутриклеточной концентрации

Возрастные иммунодефициты:
1. Иммунодефицит раннего постнатального периода.
Связан с тем, что формирование иммунной
Возрастные иммунодефициты:
1. Иммунодефицит раннего постнатального периода.
Связан с тем, что формирование иммунной

Недостаточность ИФНγ влияет на функцию макрофагов, а низкая секреторная активность Th2
Недостаточность ИФНγ влияет на функцию макрофагов, а низкая секреторная активность Th2

Собственный синтез IgG появляется к 6 месяцам, полного развития достигает к
Собственный синтез IgG появляется к 6 месяцам, полного развития достигает к

Возрастные иммунодефициты:
2. Старение иммунной системы и связанный с ним иммунодефицит.
Изменения в
Возрастные иммунодефициты:
2. Старение иммунной системы и связанный с ним иммунодефицит.
Изменения в

Основные проявление возрастных изменений тимусзависимой системы:
а) передача функции тимуса на периферию.
Основные проявление возрастных изменений тимусзависимой системы:
а) передача функции тимуса на периферию.

б) Снижение способности тимуса привлекать клетки – предшественники и его «пропускающей
б) Снижение способности тимуса привлекать клетки – предшественники и его «пропускающей

д) Функциональная недостаточность периферических Т-кл., из-за дефицита гормонов тимуса.
е) снижение численности
д) Функциональная недостаточность периферических Т-кл., из-за дефицита гормонов тимуса.
е) снижение численности

Ослабление иммунной защиты в основном затрагивает реакции, обусловленные Т-клетками, хотя явного
Ослабление иммунной защиты в основном затрагивает реакции, обусловленные Т-клетками, хотя явного

Процесс старения иммунной системы может быть ускорен неблагоприятными факторами среда. Иммунологические
Процесс старения иммунной системы может быть ускорен неблагоприятными факторами среда. Иммунологические
 Влияние цвета на человека. Цветотерапия
Влияние цвета на человека. Цветотерапия Тактика лечения хронического тонзиллита
Тактика лечения хронического тонзиллита Определение арт-терапии
Определение арт-терапии Нефротический синдром
Нефротический синдром Роль среднего медицинского персонала в профилактике нагноения послеоперационных ран
Роль среднего медицинского персонала в профилактике нагноения послеоперационных ран Диагностика беременности и бесплодия
Диагностика беременности и бесплодия Балалар тыныс алу жүйесінің патофизиологиясы
Балалар тыныс алу жүйесінің патофизиологиясы Переломы костей таза
Переломы костей таза Осложнения острого инфаркта миокарда
Осложнения острого инфаркта миокарда Массивная кровопотеря. Современные подходы к интенсивной терапии
Массивная кровопотеря. Современные подходы к интенсивной терапии Повреждения бедра и коленного сустава
Повреждения бедра и коленного сустава Пластика стебельчатым лоскутом. Лекция для студентов стоматологического факультета
Пластика стебельчатым лоскутом. Лекция для студентов стоматологического факультета Государственная политика в области иммунопрофилактики инфекционных болезней
Государственная политика в области иммунопрофилактики инфекционных болезней Бір фазалы және екі фазалы қалып алу әдістері
Бір фазалы және екі фазалы қалып алу әдістері Клещевой и центральноевропейский энцефалиты
Клещевой и центральноевропейский энцефалиты Ecology of microorganisms
Ecology of microorganisms Правила оказание первой медицинской помощи. Кровотечения
Правила оказание первой медицинской помощи. Кровотечения Лучевая терапия опухолевых и неопухолевых заболеваний
Лучевая терапия опухолевых и неопухолевых заболеваний Организация акушерско-гинекологической помощи в Украине
Организация акушерско-гинекологической помощи в Украине Методы рентгенологического исследования желудочно_кишечного тракта
Методы рентгенологического исследования желудочно_кишечного тракта Организационно –методические особенности реабилитации больных с различной патологией средствами кинезитерапии
Организационно –методические особенности реабилитации больных с различной патологией средствами кинезитерапии Сердечно-легочная реанимация. Особенности СЛР при утоплении и электротравме
Сердечно-легочная реанимация. Особенности СЛР при утоплении и электротравме Внутрибольничная инфекция
Внутрибольничная инфекция Эффективность ингаляционных кортикостероидов по сравнению с дексаметазоном в/в в лечении бронхолегочной дисплазии
Эффективность ингаляционных кортикостероидов по сравнению с дексаметазоном в/в в лечении бронхолегочной дисплазии Острые заболевания органов дыхания
Острые заболевания органов дыхания Дәріханалық шығындар, түрлері және оны болжау
Дәріханалық шығындар, түрлері және оны болжау Четыре краеугольных камня - руководства ВОЗ по планированию семьи
Четыре краеугольных камня - руководства ВОЗ по планированию семьи Дифференциальная диагностика желтушного синдрома
Дифференциальная диагностика желтушного синдрома