- Тубулопатии. Классификации

Содержание
- 2. Тубулопатии - это гетерогенная группа наследственных и приобретенных почечных канальцевых (ренальных тубулярных) дисфункций, имеющих различное течение
- 3. В основе тубулопатий лежит нарушение клеточно-специфических транспортных систем в проксимальных, дистальных канальцах и собирательных трубках нефрона.
- 4. Классификация: 1.Первичные и вторичные; 2.Наследственные и приобретенные; 3.С локализацией дефекта систем канальцевого транспорта- проксимальные, дистальные, проксимальные
- 5. Классификация тубулопатий по локализации дефекта (Ю.Е.Вельтищев, Э.А.Юрьева 1978г.)
- 6. Классификация тубулопатий в зависимости от ведущих синдромов (Ю.Е.Вельтищев, Э.А.Юрьева 1978г.)
- 7. А.В.Папаян, В.В.Красильников (1997г.): 1)Первичные тубулопатии с преимущественным поражением проксимальных извитых канальцев 2)Первичные тубулопатии с преимущественным поражением
- 8. Аминоацидурия - увеличение экскреции с мочой аминокислот. Обусловлены дефектом транспорта аминокислот в проксимальных канальцах почек. Классификация
- 9. 1.Цистинурия-аутосомно-рецессивная ацидурия, обусловленная нарушением системы клеточного транспорта в проксимальных канальцах и повышенной экскрецией с мочой цистина,
- 10. Гистидинурия - аутосомно-рецессивное. Задержка умственного развития. Метионинурия - аутосомно-рецессивное. Задержка умственного развития и эписиндром. Болезнь Хартнупа
- 11. 3.Аминоацидурия двуосновных аминокислот II типа -аутосомно-рецессивное заболевания, характеризующееся нарушенной абсорбцией двуосновных аминокислот(лизин, аргинин). Клиника: анорексия, рвота,
- 13. Почечная глюкозурия - наследственная тубулопатия, обусловленная нарушением системы транспорта глюкозы в проксимальный канальцах, проявляющаяся глюкозурией без
- 15. Типы ренальной глюкозурии: 1)Тип А - "классическая ренальная глюкозурия", при котором снижен максимальный уровень реабсорбции в
- 16. Клиника: Изолированная почечная глюкозурия - бессимптомно. Глюкозурию диагностирую обычно в грудном, раннем и дошкольном возрасте на
- 17. Диагностика Суточной глюкозурии и галактоземии -Тест на фруктозурию, аминоацидурию и галактоземию -Определить уровень глюкозы в крови
- 18. Гипофосфатемический рахит -фосфат диабет наследственная тубулопатия, характеризующаяся нарушением систем транспорта фосфатов в проксимальных канальцах почек. 3
- 19. 1)Гипофосфатемический рахит Х-сцепленный доминантный Характеризуется нарушением транспорта фосфатов в проксимальных канальцах. Ведущие симптомы: ■ Фосфатурия ■
- 20. Возможно развитие вторичного дистального почечного канальцевого ацидоза 1 типа в случаях отсутствия терапии фосфатным буфером и
- 22. 2)Аутосомно-доминантный гипофосфатемический рахит Обусловлен дефектом транспорта фосфатов в проксимальных канальцах почек, фосфатурией и гипофосфатемическим рахитом, ассоциированным
- 23. 3)Аутосомно-рецессивный гипофосфатемический рахит с гиперкальциурией Характеризуется: снижением реабсорбции фосфатов в проксимальных канальцах нефронов, проявляется гиперфосфатурией, гиперкальциурией,
- 24. Лечение предусматривает назначение активных метаболитов витамина D3, фосфатов и хирургическую ортопедическую коррекцию. ■ Фосфатный буфер (фосфат
- 25. Синдром Де Тони-Дебре-Фанкони Характеризуется клинико-лабораторным симптомокомплексом : -фосфатурия, кальциурия , глюкозурия -полидипсия, полиурия -гипераминоацидурия -гипофосфатемический рахит
- 26. Этиология: I. Наследственный 1. Идиопатический 2. Цистиноз 3. Болезнь Вильсона 4. Синдром Lowe – окуло-церебро- ренальный
- 27. Патогенез: Нарушение функции почечных проксимальных канальцев , неселективный дефект систем транспорта аминокислот, глюкозы, фосфатов и бикарбонатов.
- 28. 3. Коррекция почечного канальцевого ацидоза - бикарбонат натрия 4. Гипофосфатемический рахит -Препараты кальция -Фосфатный буфер постоянно
- 29. Почечный канальцевый метаболический ацидоз - тубулопатия , возникающая в результате дефекта реабсорбции бикарбонатов в проксимальных извитых
- 30. Аутосомно-доминантный дистальный ПКА 1 типа - классическая форма , синдром Батлера-Олбрайта - в основе которой лежит
- 31. Аутосомно-рецессивный дистальный ПКА 1 тип с глухотой Генетика: Мутации гена АТР6В1. Клиника аналогична классическому. Нейросенсорную тугоухость
- 32. Проксимальный ПКА 2 типа. Аутосомно-доминантный проксимальный ПКА 2 тип: Генетика. Дефект гена SLC9A3/ Клиника. Заболевание проявляется
- 33. Первичный проксимальный почечный ацидоз 2 типа (спорадический) объясняют незрелостью NHE-Na+H- обменивателя в проксимальных канальцах. Клиника. В
- 34. ПКА 4 типа с гиперкалиемией Первичный гиперкалиемический ПКА 4 типа раннего детства (транзиторный) проявляется у младенцев
- 35. Псевдогипоальдостеронизм - гетерогенное наследственное заболевание, обусловленное резистентностью или дефицитом рецепторов эпителия канальцев к альдостерону, что приводит
- 36. 1 тип. 3 вида ренального ПГА: · Вариант 1-почечный ПГА 1 типа классический аут-дом · Вариант
- 37. 1 тип: AR (аут-рец): почки: потеря соли, гипонатриемия, гиперкалиемия, метаболический ацидоз, повышение концентрации альдостерона и активности
- 38. 2 тип A-D (аут-дом): гиперкалиемия, гипертензия, гиперхлоремический ацидоз, норм.уровень концентрации альдостерона плазмы, снижение активности ренина плазмы.
- 39. Тубулопатии с ведущим синдромом алкалоза 1.Синдром Гительмана 2.Синдром Барттера 3.Синдром Лиддла
- 40. Синдром Гительмана (Gitelman Syndrome) - это наследственная тубулопатия с аутосомно-рецессивным типом наследования, описанная в 1966 году
- 41. Патогенез
- 42. Клиника Первые признаки диагностируется чаще в дошкольном возрасте. Проявляется гипокалиемией, мышечной гипотонией, гипомагнезиемией, метаболическим алкалозом, нормокальциемией,
- 43. Лечение Коррекция гипокалиемии - препараты калия ( аспаркам, панангин, хлорид калия) Препараты магния для снижения риска
- 44. Синдром Барттера – гетерогенная тубулопатия с нарушением систем транспорта K, Na, Cl в дистальном канальце в
- 45. Патогенез
- 46. Антенатальный синдром Барттера Может быть диагностирован in utero: необъяснимое многоводие между 24 и 36 неделями нормальный
- 47. Лечение антенатального синдрома Барттера Заместительная терапия после родов: немедленное введение раствора NaCl потеря воды – до
- 48. Синдром Барттера III типа Клиника: нефрокальциноз отсутствует, выраженная гипокалиемия обусловливает изменения в миокарде, нарушение ритма сердца.
- 49. Синдром Лиддла - наследственная тубулопатия с аутосомно-доминантным типом наследования, характеризуется расстройством почечно-клеточного транспорта, клинически напоминает первичный
- 50. Клиника : -АГ -Полиурия -Гипокалиемия -Метаболический алкалоз. Диагностика: -Уровень натрия в моче(менее 20 мЭкв). -Активность ренина
- 51. ТУБУЛОПАТИИ С ВЕДУЩИМ СИДРОМОМ НЕФРОКАЛЬЦИНОЗА 1. СЕМЕЙНАЯ ГИПОМАГНИЕМИЯ - ГИПЕРКАЛЬЦИУРИЯ С НЕФРОКАЛЬЦИНОЗОМ 2. ПЕРВИЧНАЯ ГИПЕРОКСАЛУРИЯ 1
- 52. 1. СЕМЕЙНАЯ ГИПОМАГНИЕМИЯ - ГИПЕРКАЛЬЦИУРИЯ С НЕФРОКАЛЬЦИНОЗОМ — тубулопатия с аутосомно-доминантным типом наследования, часто прогрессирующая в
- 53. Выделяют 4 варианта снижения Mg2+ в крови (наследственной гипомагниемии): * семейную гипомагниемию сгиперкальциурией и нефрокальцинозом (с
- 54. Клиника. Заболевание проявляется у детей в грудном и раннем возрасте. Характерны полиурия, полидипсия. При обследовании пациентов
- 55. 2.Первичная гипероксалурия 1 типа - аутосомно-рецессивное заболевание, хар-ся повышенными продукцией оксалата в пероксисомах и экскреции оксалатов
- 56. Клиника. Для первичной гипероксалурии 1 типа у детей раннего возраста хар-ны повышение экскреции оксалатов с мочой,
- 57. Синдром Дента -Х-сцепленный рецессивный гипофосфатемический рахит. Характеризуется: · протеинурией; · нефрокальцинозом; · синдромом Фанкони. Генетика. Тип
- 58. Экскреция с мочой цитратов, мочевой кислоты, оксалатов нормальная , характерна повышенная экскреция кальция до 10-20 мг/кг/сут
- 59. Лабораторные исследования: нефрокальциноз (75%), почечные оксалатно-кальциевые, кальций-фосфатные камни (50%). Лечение: !Важно пить много жидкости и соблюдать
- 60. Врожденный нефрогенный несахарный диабет ВННД(почечный несахарный дибет)-наследственное заболевание,ведущими симптомами которого являются полиурия,полидипсия и гипостенурия. Классификация: Первичный(наследственный,врожденный)
- 61. Причины: -лекарственный препараты -аналгетическая нефропатия -серповидно-клеточная нефропатия -гипокалиемия -гипокальциемия -обструктивная уропатия -почечная дисплазия -хронический пиелонефри -хроническая
- 63. Диагностика: I. Молекулярно-генетическая диагностика ННД II. Дифференциально-диагностические тесты концентрационной способности почек при полиурии у детей III.
- 65. Скачать презентацию

Тубулопатии - это гетерогенная группа наследственных и приобретенных почечных канальцевых (ренальных
Тубулопатии - это гетерогенная группа наследственных и приобретенных почечных канальцевых (ренальных
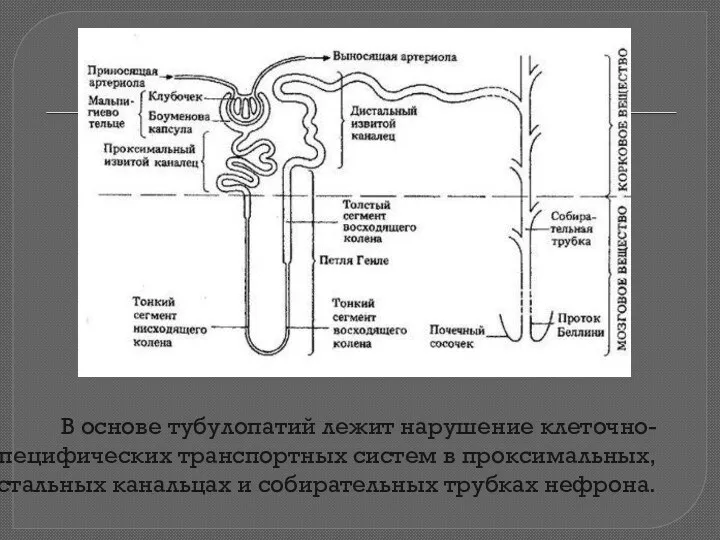
В основе тубулопатий лежит нарушение клеточно-специфических транспортных систем в проксимальных, дистальных
В основе тубулопатий лежит нарушение клеточно-специфических транспортных систем в проксимальных, дистальных

Классификация:
1.Первичные и вторичные;
2.Наследственные и приобретенные;
3.С локализацией дефекта систем канальцевого транспорта- проксимальные,
Классификация:
1.Первичные и вторичные;
2.Наследственные и приобретенные;
3.С локализацией дефекта систем канальцевого транспорта- проксимальные,

Классификация тубулопатий по локализации дефекта (Ю.Е.Вельтищев, Э.А.Юрьева 1978г.)
Классификация тубулопатий по локализации дефекта (Ю.Е.Вельтищев, Э.А.Юрьева 1978г.)

Классификация тубулопатий в зависимости от ведущих синдромов
(Ю.Е.Вельтищев, Э.А.Юрьева 1978г.)
Классификация тубулопатий в зависимости от ведущих синдромов
(Ю.Е.Вельтищев, Э.А.Юрьева 1978г.)

А.В.Папаян, В.В.Красильников (1997г.):
1)Первичные тубулопатии с преимущественным поражением проксимальных извитых канальцев
2)Первичные тубулопатии
А.В.Папаян, В.В.Красильников (1997г.):
1)Первичные тубулопатии с преимущественным поражением проксимальных извитых канальцев
2)Первичные тубулопатии

Аминоацидурия - увеличение экскреции с мочой аминокислот. Обусловлены дефектом транспорта аминокислот
Аминоацидурия - увеличение экскреции с мочой аминокислот. Обусловлены дефектом транспорта аминокислот

1.Цистинурия-аутосомно-рецессивная ацидурия, обусловленная нарушением системы клеточного транспорта в проксимальных канальцах и
1.Цистинурия-аутосомно-рецессивная ацидурия, обусловленная нарушением системы клеточного транспорта в проксимальных канальцах и
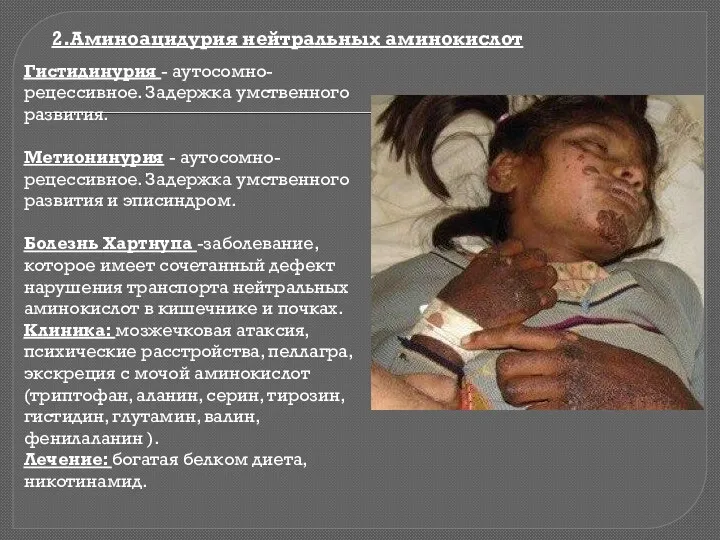
Гистидинурия - аутосомно-рецессивное. Задержка умственного развития.
Метионинурия - аутосомно-рецессивное. Задержка умственного развития
Гистидинурия - аутосомно-рецессивное. Задержка умственного развития.
Метионинурия - аутосомно-рецессивное. Задержка умственного развития

3.Аминоацидурия двуосновных аминокислот II типа -аутосомно-рецессивное заболевания, характеризующееся нарушенной абсорбцией двуосновных
3.Аминоацидурия двуосновных аминокислот II типа -аутосомно-рецессивное заболевания, характеризующееся нарушенной абсорбцией двуосновных
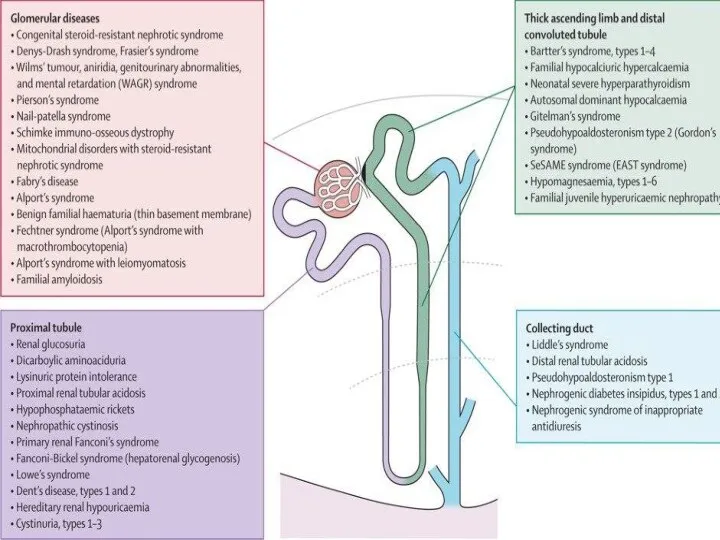
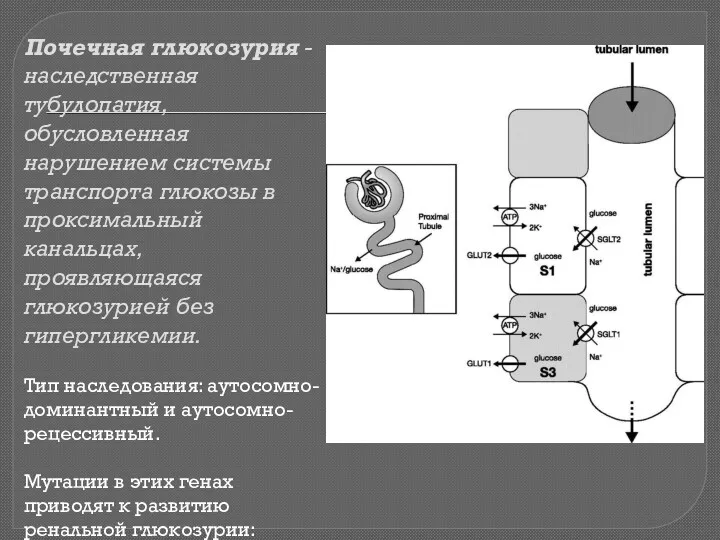
Почечная глюкозурия - наследственная тубулопатия, обусловленная нарушением системы транспорта глюкозы в
Почечная глюкозурия - наследственная тубулопатия, обусловленная нарушением системы транспорта глюкозы в
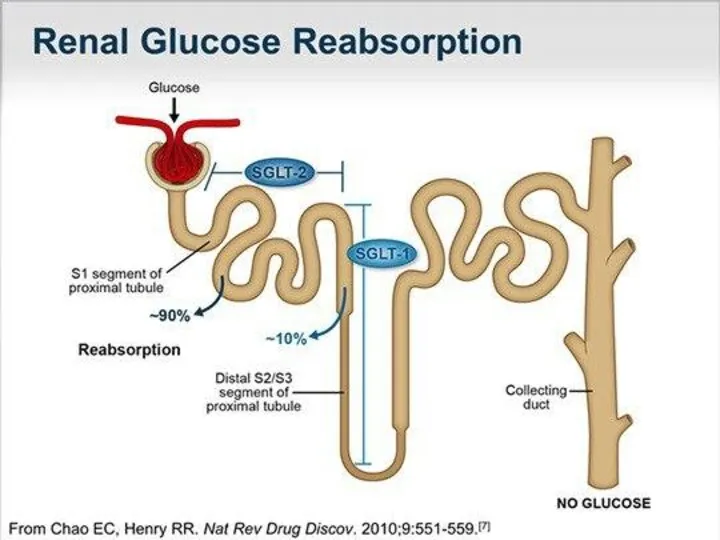

Типы ренальной глюкозурии:
1)Тип А - "классическая ренальная глюкозурия", при котором снижен
Типы ренальной глюкозурии: 1)Тип А - "классическая ренальная глюкозурия", при котором снижен

Клиника:
Изолированная почечная глюкозурия - бессимптомно.
Глюкозурию диагностирую обычно в грудном, раннем и
Клиника:
Изолированная почечная глюкозурия - бессимптомно.
Глюкозурию диагностирую обычно в грудном, раннем и

Диагностика
Суточной глюкозурии и галактоземии
-Тест на фруктозурию, аминоацидурию и галактоземию
-Определить уровень
Диагностика
Суточной глюкозурии и галактоземии
-Тест на фруктозурию, аминоацидурию и галактоземию
-Определить уровень

Гипофосфатемический рахит -фосфат диабет
наследственная тубулопатия, характеризующаяся нарушением систем транспорта фосфатов в
Гипофосфатемический рахит -фосфат диабет
наследственная тубулопатия, характеризующаяся нарушением систем транспорта фосфатов в

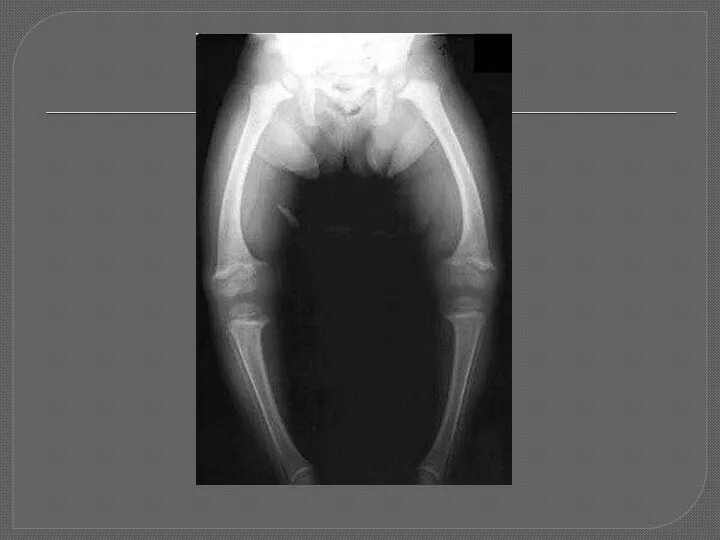
1)Гипофосфатемический рахит Х-сцепленный доминантный
Характеризуется нарушением транспорта фосфатов в проксимальных канальцах.
Ведущие симптомы:
■
1)Гипофосфатемический рахит Х-сцепленный доминантный
Характеризуется нарушением транспорта фосфатов в проксимальных канальцах.
Ведущие симптомы:
■

Возможно развитие вторичного дистального почечного канальцевого ацидоза 1 типа в
Возможно развитие вторичного дистального почечного канальцевого ацидоза 1 типа в
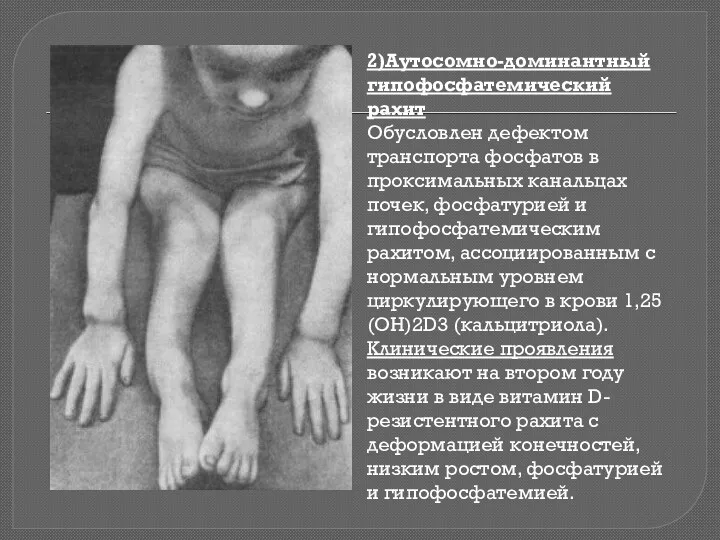
2)Аутосомно-доминантный гипофосфатемический рахит
Обусловлен дефектом транспорта фосфатов в проксимальных канальцах почек, фосфатурией
2)Аутосомно-доминантный гипофосфатемический рахит
Обусловлен дефектом транспорта фосфатов в проксимальных канальцах почек, фосфатурией
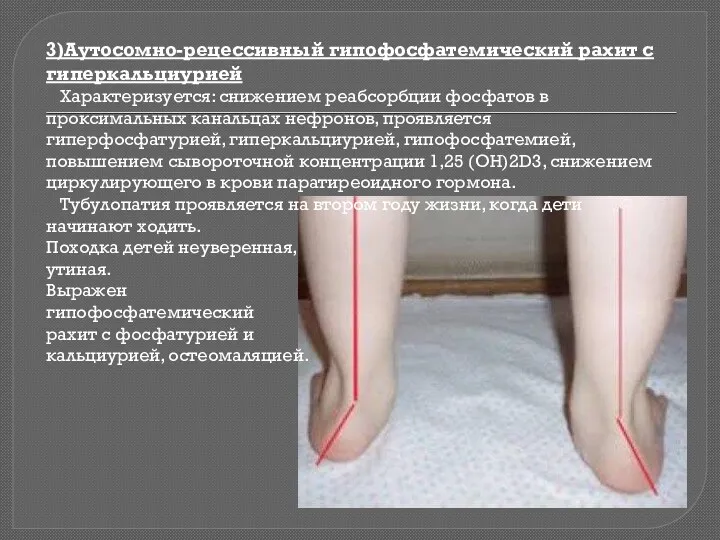
3)Аутосомно-рецессивный гипофосфатемический рахит с гиперкальциурией
Характеризуется: снижением реабсорбции фосфатов в проксимальных
3)Аутосомно-рецессивный гипофосфатемический рахит с гиперкальциурией
Характеризуется: снижением реабсорбции фосфатов в проксимальных

Лечение предусматривает назначение активных метаболитов витамина D3, фосфатов и хирургическую ортопедическую
Лечение предусматривает назначение активных метаболитов витамина D3, фосфатов и хирургическую ортопедическую

Синдром Де Тони-Дебре-Фанкони
Характеризуется клинико-лабораторным симптомокомплексом :
-фосфатурия, кальциурия , глюкозурия
-полидипсия, полиурия
-гипераминоацидурия
-гипофосфатемический рахит
Синдром Де Тони-Дебре-Фанкони Характеризуется клинико-лабораторным симптомокомплексом : -фосфатурия, кальциурия , глюкозурия -полидипсия, полиурия -гипераминоацидурия -гипофосфатемический рахит

Этиология:
I. Наследственный
1. Идиопатический
2. Цистиноз
3. Болезнь Вильсона
4. Синдром Lowe – окуло-церебро- ренальный
5.
Этиология: I. Наследственный 1. Идиопатический 2. Цистиноз 3. Болезнь Вильсона 4. Синдром Lowe – окуло-церебро- ренальный 5.

Патогенез:
Нарушение функции почечных проксимальных канальцев , неселективный дефект систем транспорта аминокислот,
Патогенез: Нарушение функции почечных проксимальных канальцев , неселективный дефект систем транспорта аминокислот,

3. Коррекция почечного канальцевого ацидоза - бикарбонат натрия
4. Гипофосфатемический рахит
-Препараты кальция
-Фосфатный
3. Коррекция почечного канальцевого ацидоза - бикарбонат натрия 4. Гипофосфатемический рахит -Препараты кальция -Фосфатный

Почечный канальцевый метаболический ацидоз - тубулопатия , возникающая в результате дефекта
Почечный канальцевый метаболический ацидоз - тубулопатия , возникающая в результате дефекта

Аутосомно-доминантный дистальный ПКА 1 типа - классическая форма , синдром Батлера-Олбрайта
Аутосомно-доминантный дистальный ПКА 1 типа - классическая форма , синдром Батлера-Олбрайта

Аутосомно-рецессивный дистальный ПКА 1 тип с глухотой
Генетика: Мутации гена АТР6В1. Клиника
Аутосомно-рецессивный дистальный ПКА 1 тип с глухотой
Генетика: Мутации гена АТР6В1. Клиника

Проксимальный ПКА 2 типа.
Аутосомно-доминантный проксимальный ПКА 2 тип:
Генетика. Дефект гена SLC9A3/
Клиника.
Проксимальный ПКА 2 типа.
Аутосомно-доминантный проксимальный ПКА 2 тип:
Генетика. Дефект гена SLC9A3/
Клиника.

Первичный проксимальный почечный ацидоз 2 типа (спорадический) объясняют незрелостью NHE-Na+H- обменивателя
Первичный проксимальный почечный ацидоз 2 типа (спорадический) объясняют незрелостью NHE-Na+H- обменивателя

ПКА 4 типа с гиперкалиемией
Первичный гиперкалиемический ПКА 4 типа раннего детства
ПКА 4 типа с гиперкалиемией
Первичный гиперкалиемический ПКА 4 типа раннего детства

Псевдогипоальдостеронизм - гетерогенное наследственное заболевание, обусловленное резистентностью или дефицитом рецепторов эпителия
Псевдогипоальдостеронизм - гетерогенное наследственное заболевание, обусловленное резистентностью или дефицитом рецепторов эпителия

1 тип. 3 вида ренального ПГА:
· Вариант 1-почечный ПГА 1 типа
1 тип. 3 вида ренального ПГА:
· Вариант 1-почечный ПГА 1 типа

1 тип: AR (аут-рец): почки: потеря соли, гипонатриемия, гиперкалиемия, метаболический ацидоз,
1 тип: AR (аут-рец): почки: потеря соли, гипонатриемия, гиперкалиемия, метаболический ацидоз,

2 тип A-D (аут-дом): гиперкалиемия, гипертензия, гиперхлоремический ацидоз, норм.уровень концентрации альдостерона
2 тип A-D (аут-дом): гиперкалиемия, гипертензия, гиперхлоремический ацидоз, норм.уровень концентрации альдостерона

Тубулопатии с ведущим синдромом алкалоза
1.Синдром Гительмана
2.Синдром Барттера
3.Синдром Лиддла
Тубулопатии с ведущим синдромом алкалоза
1.Синдром Гительмана
2.Синдром Барттера
3.Синдром Лиддла

Синдром Гительмана (Gitelman Syndrome) - это наследственная тубулопатия с аутосомно-рецессивным типом
Синдром Гительмана (Gitelman Syndrome) - это наследственная тубулопатия с аутосомно-рецессивным типом
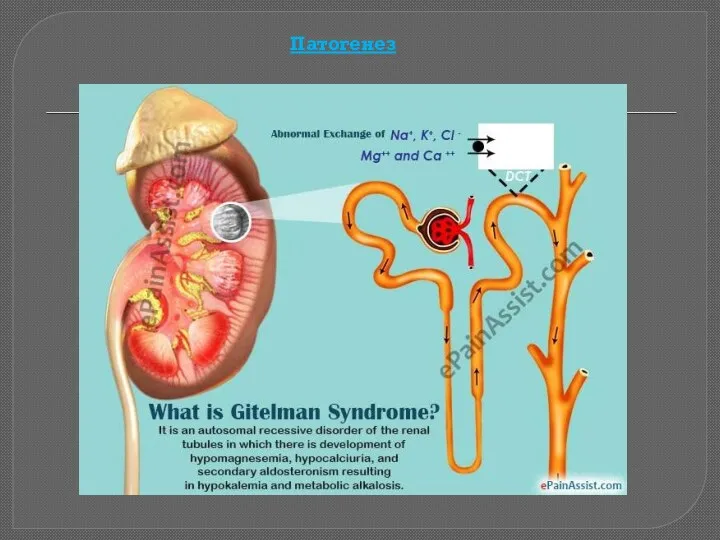
Патогенез
Патогенез
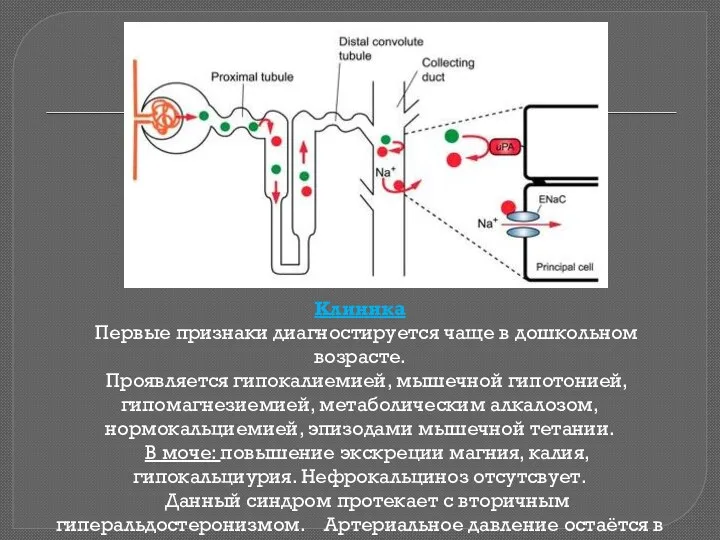
Клиника
Первые признаки диагностируется чаще в дошкольном возрасте.
Проявляется гипокалиемией, мышечной
Клиника Первые признаки диагностируется чаще в дошкольном возрасте. Проявляется гипокалиемией, мышечной

Лечение
Коррекция гипокалиемии - препараты калия ( аспаркам, панангин, хлорид калия)
Препараты магния
Лечение Коррекция гипокалиемии - препараты калия ( аспаркам, панангин, хлорид калия) Препараты магния

Синдром Барттера – гетерогенная тубулопатия с нарушением систем транспорта K, Na,
Синдром Барттера – гетерогенная тубулопатия с нарушением систем транспорта K, Na,
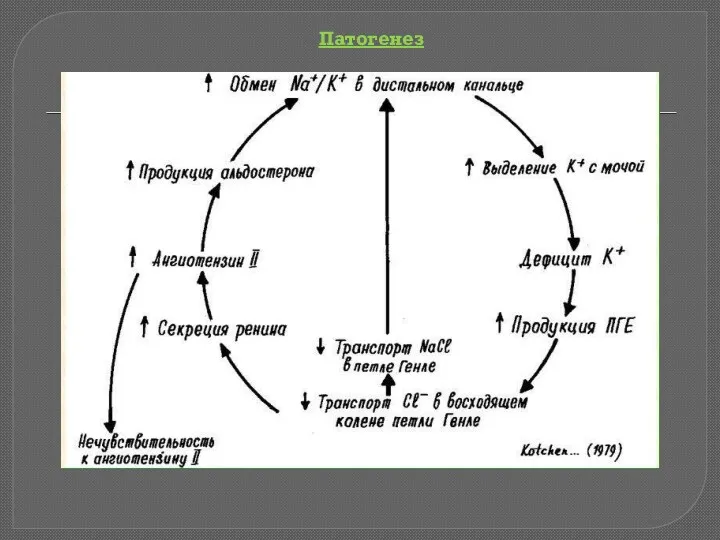
Патогенез
Патогенез

Антенатальный синдром Барттера
Может быть диагностирован in utero: необъяснимое многоводие между 24
Антенатальный синдром Барттера
Может быть диагностирован in utero: необъяснимое многоводие между 24

Лечение антенатального синдрома Барттера
Заместительная терапия после родов:
немедленное введение раствора NaCl потеря
Лечение антенатального синдрома Барттера
Заместительная терапия после родов:
немедленное введение раствора NaCl потеря

Синдром Барттера III типа
Клиника: нефрокальциноз отсутствует, выраженная гипокалиемия обусловливает изменения в
Синдром Барттера III типа
Клиника: нефрокальциноз отсутствует, выраженная гипокалиемия обусловливает изменения в

Синдром Лиддла - наследственная тубулопатия с аутосомно-доминантным типом наследования, характеризуется расстройством
Синдром Лиддла - наследственная тубулопатия с аутосомно-доминантным типом наследования, характеризуется расстройством

Клиника :
-АГ
-Полиурия
-Гипокалиемия
-Метаболический алкалоз.
Диагностика:
-Уровень натрия в моче(менее 20 мЭкв).
-Активность ренина плазмы и
Клиника :
-АГ
-Полиурия
-Гипокалиемия
-Метаболический алкалоз.
Диагностика:
-Уровень натрия в моче(менее 20 мЭкв).
-Активность ренина плазмы и

ТУБУЛОПАТИИ С ВЕДУЩИМ СИДРОМОМ НЕФРОКАЛЬЦИНОЗА
1. СЕМЕЙНАЯ ГИПОМАГНИЕМИЯ - ГИПЕРКАЛЬЦИУРИЯ С НЕФРОКАЛЬЦИНОЗОМ
2.
ТУБУЛОПАТИИ С ВЕДУЩИМ СИДРОМОМ НЕФРОКАЛЬЦИНОЗА 1. СЕМЕЙНАЯ ГИПОМАГНИЕМИЯ - ГИПЕРКАЛЬЦИУРИЯ С НЕФРОКАЛЬЦИНОЗОМ 2.

1. СЕМЕЙНАЯ ГИПОМАГНИЕМИЯ - ГИПЕРКАЛЬЦИУРИЯ С НЕФРОКАЛЬЦИНОЗОМ
— тубулопатия с аутосомно-доминантным типом
1. СЕМЕЙНАЯ ГИПОМАГНИЕМИЯ - ГИПЕРКАЛЬЦИУРИЯ С НЕФРОКАЛЬЦИНОЗОМ — тубулопатия с аутосомно-доминантным типом

Выделяют 4 варианта снижения Mg2+ в крови (наследственной гипомагниемии):
* семейную гипомагниемию
Выделяют 4 варианта снижения Mg2+ в крови (наследственной гипомагниемии):
* семейную гипомагниемию

Клиника.
Заболевание проявляется у детей в грудном и раннем возрасте. Характерны полиурия,
Клиника. Заболевание проявляется у детей в грудном и раннем возрасте. Характерны полиурия,

2.Первичная гипероксалурия 1 типа - аутосомно-рецессивное заболевание, хар-ся повышенными продукцией оксалата
2.Первичная гипероксалурия 1 типа - аутосомно-рецессивное заболевание, хар-ся повышенными продукцией оксалата

Клиника.
Для первичной гипероксалурии 1 типа у детей раннего возраста хар-ны повышение
Клиника. Для первичной гипероксалурии 1 типа у детей раннего возраста хар-ны повышение

Синдром Дента -Х-сцепленный рецессивный гипофосфатемический рахит. Характеризуется:
· протеинурией;
· нефрокальцинозом;
· синдромом Фанкони.
Синдром Дента -Х-сцепленный рецессивный гипофосфатемический рахит. Характеризуется:
· протеинурией;
· нефрокальцинозом;
· синдромом Фанкони.

Экскреция с мочой цитратов, мочевой кислоты, оксалатов нормальная , характерна повышенная
Экскреция с мочой цитратов, мочевой кислоты, оксалатов нормальная , характерна повышенная

Лабораторные исследования: нефрокальциноз (75%), почечные оксалатно-кальциевые, кальций-фосфатные камни (50%).
Лечение:
!Важно пить
Лабораторные исследования: нефрокальциноз (75%), почечные оксалатно-кальциевые, кальций-фосфатные камни (50%).
Лечение:
!Важно пить

Врожденный нефрогенный несахарный диабет ВННД(почечный несахарный дибет)-наследственное заболевание,ведущими симптомами которого являются
Врожденный нефрогенный несахарный диабет ВННД(почечный несахарный дибет)-наследственное заболевание,ведущими симптомами которого являются

Причины:
-лекарственный препараты
-аналгетическая нефропатия
-серповидно-клеточная нефропатия
-гипокалиемия
-гипокальциемия
-обструктивная уропатия
-почечная дисплазия
-хронический пиелонефри
-хроническая уремическая нефропатия.
-амилоидоз ,саркоидоз
Вторичный
Причины:
-лекарственный препараты
-аналгетическая нефропатия
-серповидно-клеточная нефропатия
-гипокалиемия
-гипокальциемия
-обструктивная уропатия
-почечная дисплазия
-хронический пиелонефри
-хроническая уремическая нефропатия.
-амилоидоз ,саркоидоз
Вторичный


Диагностика:
I. Молекулярно-генетическая диагностика ННД
II. Дифференциально-диагностические тесты концентрационной способности почек при полиурии
Диагностика:
I. Молекулярно-генетическая диагностика ННД
II. Дифференциально-диагностические тесты концентрационной способности почек при полиурии
 Гіподинамія – ворог сучасної людини
Гіподинамія – ворог сучасної людини Жарақаттар
Жарақаттар Синдром гипотиреоза
Синдром гипотиреоза Ерте кезеңдегі токсикоз
Ерте кезеңдегі токсикоз Артериальные гипертензии
Артериальные гипертензии Возможности и ограничения личности при выборе профессиональной деятельности. Здоровье человека и выбор профессии
Возможности и ограничения личности при выборе профессиональной деятельности. Здоровье человека и выбор профессии Осложнения язвенной болезни
Осложнения язвенной болезни Что такое ВИЧ, СПИД. (10 класс)
Что такое ВИЧ, СПИД. (10 класс) 170 years since the birth of Ivan Pavlov. Traditions and innovations in Cognitive Behavioral Therapy
170 years since the birth of Ivan Pavlov. Traditions and innovations in Cognitive Behavioral Therapy Сибирская язва
Сибирская язва Сестринская практика, основанная на принципах доказательной медицины
Сестринская практика, основанная на принципах доказательной медицины Сосудистые заболевания головного мозга
Сосудистые заболевания головного мозга Трансплантационный иммунитет. Иммунологические аспекты переливания крови
Трансплантационный иммунитет. Иммунологические аспекты переливания крови Биомеханика и механика. Опора
Биомеханика и механика. Опора Нейротропные средства
Нейротропные средства Местное обезболивание
Местное обезболивание Клинический случай (плановый)
Клинический случай (плановый) Дія електричного струму на організм людини. Електротравма. Надання домедичної допомоги при ураженні електричним струмом
Дія електричного струму на організм людини. Електротравма. Надання домедичної допомоги при ураженні електричним струмом Принципы организации первичной медико-санитарной помощи
Принципы организации первичной медико-санитарной помощи Pathophysiology of diabetes mellitus. Specific forms of diabetes
Pathophysiology of diabetes mellitus. Specific forms of diabetes Респираторная поддержка при межгоспитальной транспортировке больных с тяжелой сочетанной травмой и нарушением витальных функций
Респираторная поддержка при межгоспитальной транспортировке больных с тяжелой сочетанной травмой и нарушением витальных функций Организация службы скорой медицинской помощи
Организация службы скорой медицинской помощи Жидкие лекарственные формы. Лекция 3
Жидкие лекарственные формы. Лекция 3 Анемия. Морфологическая классификация
Анемия. Морфологическая классификация Бронхиальная астма
Бронхиальная астма Методы полногеномного анализа в медицине. Курс 3 ЦИОП Медицина будущего
Методы полногеномного анализа в медицине. Курс 3 ЦИОП Медицина будущего Рентген диагностика ахалазия кардии
Рентген диагностика ахалазия кардии Дифференциальная диагностика острой хирургической патологии у детей
Дифференциальная диагностика острой хирургической патологии у детей