Болезнь Шарко-Мари-Тута Наследственная моторно-сенсорная нейропатия невральная амиотрофия презентация
- Болезнь Шарко-Мари-Тута Наследственная моторно-сенсорная нейропатия невральная амиотрофия

Содержание
- 2. Болезнь Шарко–Мари–Тута (ШМТ) – это обширная группа наследственных болезней нервной системы, характеризующихся хронически прогрессирующей слабостью и
- 3. Наследственное заболвание нервов (невропатия), которое принимает различные формы. На данный момент является неизлечимым. является одним из
- 4. В основе болезни лежит дегенерация двига тельных и чувствительных волокон пе риферических нервов Манифестация симптомов обычно
- 5. По ЭНМГ и гистопатологическим критериям различают два основных типа ШМТ: миелинопатии – ШМТ типа 1 (ШМТ1,
- 6. Для ШМТ1 характерно существенное снижение скорости проведения импульса (СПИ) по периферическим нервам вследствие сегментарной де- и
- 7. ШМТ3 типа БОЛЕЗНЬ ДЕЖЕРИНА-СОТТА Очень редко Аутосомно-доминантный и АР Причина – демиелинизация Описаны мутации в 4х
- 8. Слабость и снижении чувствительнгости в конечностях Утрата мышечной массы и сухожильных рефлексов Боль в конечностях Искривление
- 9. Гипертрофия подкожных нервов( за счет разрастания соединительной ткани): Малоберцового Большого ушного
- 10. Для 1 и 2 формы Дебют симптомов отмечается у мужчин в 15–20 летнем возрасте, а у
- 11. Типичный̆ симптомокомплекс включает: симметричную прогрессирующую слабость и атрофию мышц стоп, голеней̆, нижней̆ трети бедер; снижение и
- 12. деформацию стоп по типу “эквиноварусных” и, в более поздней стадии, кистей рук (“обезьянья лапа”); изменение походки,
- 13. Инвалидизация у всех пациентов наступает через 15–20 лет после на чала заболевания. Некоторые больные сохраняют способность
- 14. Диагностика на основе жалоб больного, клинических проявлений, собранного анамнеза, данных лабораторного и инструментального обследования. Подтверждение диагноза
- 15. Дифдиагностика проводят с миопатией Говерса–Веландера, спинальной амиотрофией, нейропатией полиневритами
- 16. Лечение Хотя в настоящее время нет эффективного лечения расстройства, однако было предложено использование аскорбиновой кислоты является
- 18. Скачать презентацию

Болезнь Шарко–Мари–Тута (ШМТ) – это обширная группа наследственных болезней нервной системы,
Болезнь Шарко–Мари–Тута (ШМТ) – это обширная группа наследственных болезней нервной системы,

Наследственное заболвание нервов (невропатия), которое принимает различные формы.
На данный момент
Наследственное заболвание нервов (невропатия), которое принимает различные формы.
На данный момент

В основе болезни лежит дегенерация двига тельных и чувствительных волокон пе
В основе болезни лежит дегенерация двига тельных и чувствительных волокон пе

По ЭНМГ и гистопатологическим критериям различают два основных типа ШМТ:
миелинопатии
По ЭНМГ и гистопатологическим критериям различают два основных типа ШМТ:
миелинопатии

Для ШМТ1 характерно существенное снижение скорости проведения импульса (СПИ) по периферическим
Для ШМТ1 характерно существенное снижение скорости проведения импульса (СПИ) по периферическим

ШМТ3 типа
БОЛЕЗНЬ ДЕЖЕРИНА-СОТТА
Очень редко
Аутосомно-доминантный и АР
Причина – демиелинизация
Описаны мутации в
ШМТ3 типа
БОЛЕЗНЬ ДЕЖЕРИНА-СОТТА
Очень редко
Аутосомно-доминантный и АР
Причина – демиелинизация
Описаны мутации в

Слабость и снижении чувствительнгости в конечностях
Утрата мышечной массы и сухожильных рефлексов
Боль
Слабость и снижении чувствительнгости в конечностях
Утрата мышечной массы и сухожильных рефлексов
Боль

Гипертрофия подкожных нервов( за счет разрастания соединительной ткани):
Малоберцового
Большого ушного
Гипертрофия подкожных нервов( за счет разрастания соединительной ткани):
Малоберцового
Большого ушного

Для 1 и 2 формы
Дебют симптомов отмечается у мужчин в 15–20
Для 1 и 2 формы
Дебют симптомов отмечается у мужчин в 15–20
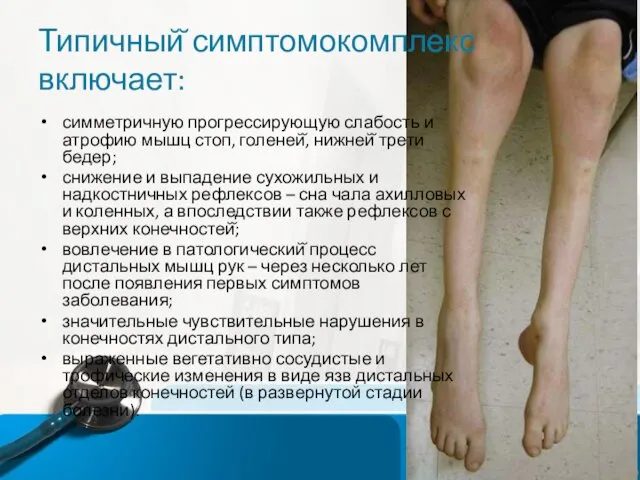
Типичный̆ симптомокомплекс включает:
симметричную прогрессирующую слабость и атрофию мышц стоп, голеней̆, нижней̆
Типичный̆ симптомокомплекс включает:
симметричную прогрессирующую слабость и атрофию мышц стоп, голеней̆, нижней̆
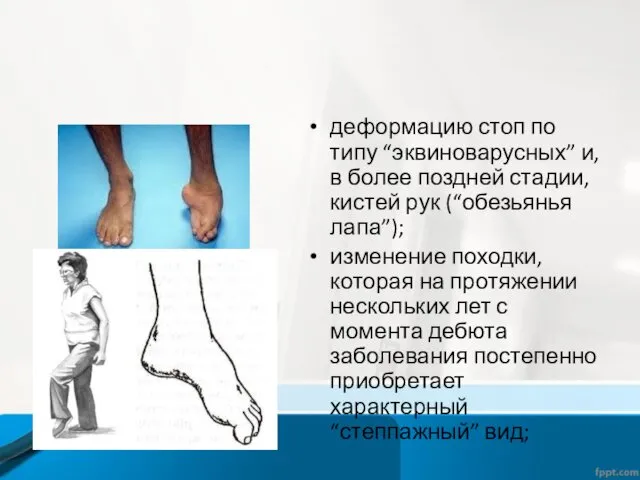
деформацию стоп по типу “эквиноварусных” и, в более поздней стадии, кистей
деформацию стоп по типу “эквиноварусных” и, в более поздней стадии, кистей

Инвалидизация у всех пациентов наступает через 15–20 лет после на чала
Инвалидизация у всех пациентов наступает через 15–20 лет после на чала

Диагностика
на основе жалоб больного, клинических проявлений, собранного анамнеза, данных лабораторного и
Диагностика
на основе жалоб больного, клинических проявлений, собранного анамнеза, данных лабораторного и

Дифдиагностика
проводят с миопатией Говерса–Веландера,
спинальной амиотрофией,
нейропатией
полиневритами
Дифдиагностика
проводят с миопатией Говерса–Веландера,
спинальной амиотрофией,
нейропатией
полиневритами

Лечение
Хотя в настоящее время нет эффективного лечения расстройства, однако было предложено
Лечение
Хотя в настоящее время нет эффективного лечения расстройства, однако было предложено
 Административная ответственность несовершеннолетних
Административная ответственность несовершеннолетних Определение угла. Развёрнутый угол
Определение угла. Развёрнутый угол Товарный знак. Сходство до степени смешения
Товарный знак. Сходство до степени смешения Решение олимпиадных задач по математике.
Решение олимпиадных задач по математике. Кроссфит
Кроссфит Сервис Online Test Pad. Создаем интерактивные кроссворды, опросы, тесты
Сервис Online Test Pad. Создаем интерактивные кроссворды, опросы, тесты Презентация урока по ОПК Православное учение о человеке
Презентация урока по ОПК Православное учение о человеке Рождество Христово
Рождество Христово Құтадғу білік
Құтадғу білік Презентация Правильное питание
Презентация Правильное питание 20231126_moya_budushchaya_professiya_dizayner_0
20231126_moya_budushchaya_professiya_dizayner_0 Презентация к родительскому собранию УСПЕШНОСТЬ ОБУЧЕНИЯ МЛАДШЕГО ШКОЛЬНИКА, ПОМОГИ ЕМУ УЧИТЬСЯ
Презентация к родительскому собранию УСПЕШНОСТЬ ОБУЧЕНИЯ МЛАДШЕГО ШКОЛЬНИКА, ПОМОГИ ЕМУ УЧИТЬСЯ Реконструкция электрической части станции типа КЭС
Реконструкция электрической части станции типа КЭС Подготовка к эксплуатации и освоение скважин
Подготовка к эксплуатации и освоение скважин Анализ поизводственного травматизма
Анализ поизводственного травматизма Пример записи решения задания
Пример записи решения задания Карбоновые кислоты
Карбоновые кислоты Инфекционная служба в Республике Казахстан: сегодня и с внедрением ОСМС
Инфекционная служба в Республике Казахстан: сегодня и с внедрением ОСМС Милосердие, забота о слабых, взаимопомощь ОРКСЭ
Милосердие, забота о слабых, взаимопомощь ОРКСЭ Ложный круп
Ложный круп Презентация опыта работы
Презентация опыта работы Общие вопросы аттестации объектов информатизации
Общие вопросы аттестации объектов информатизации Територія оптимізму
Територія оптимізму Патогенез туберкулеза. Иммунитет и аллергия. Основы иммунодиагностики туберкулезной инфекции. Лекция 2
Патогенез туберкулеза. Иммунитет и аллергия. Основы иммунодиагностики туберкулезной инфекции. Лекция 2 Теории происхождения языка
Теории происхождения языка Оборона масса айлыгында башкарылган эшләр
Оборона масса айлыгында башкарылган эшләр Случаи вычитания 14-
Случаи вычитания 14- Nike. How the brand survived until today
Nike. How the brand survived until today