- Эпигенетика. Импринтинг и наследственная патология у человека

Содержание
- 2. Геномный импринтинг - эпигенетический механизм регуляции экспрессии гомологичных генов в процессе развития организма в зависимости от
- 3. Геномный импринтинг Эпигенетический феномен, дифференцирующий материнские и отцовские копии генов в геноме организма. Подобная дифференцировка обусловливает
- 7. Метилирование/деметилирование в процессе гаметогенеза
- 9. Стресс-активируемая рибонуклеаза Angiogenin разрезает зрелую tРНК в антикодоновой петле, что дает две половинки, которые и представляют
- 10. - Несколько сотен важных для развития малых РНК соматического происхождения доставляются в сперматозоид специальным классом нановезикул
- 11. Нановизикулы – переносчики нкРНК.
- 12. Барьер Вейсмана – миф! Tелегония на марше: (проявление признаков первого самца у потомства в животном мире)
- 13. Tелегония В XIX в. лорд Мортон, близкий друг Ч. Дарвина отважился на биологический опыт: скрестил чистопородную
- 14. Для нормального развития необходим равный вклад обоих родителей. Трансплантация пронуклеусов. Андрогенетические зиготы - нормальное развитие зародышевых
- 16. Наши основоположники В.С. Баранов А.П. Дыбан, 1922-2002 А.П. Дыбан и В.С.Баранов внесли значительный вклад в экспериментальную
- 17. Однородительская дисомия. На мышиных транслокационных гибридах, несущих отдельные хромосомные участки, имеющие как отцовское, так и материнское
- 19. Механизмы формирования ОРД у человека. 1) Комплементация гамет – дополнение нуллисомной по определенной хромосоме набора одной
- 21. ОРД по целым хромосомам или их фрагментам выявлены при анализе наследственной патологии и у человека. материнская
- 22. Предполагается, что геном человека содержит не менее 200 импринтированных генов. На сегодняшний день их около 150,
- 23. Болезни импринтинга Синдромы Прадера-Вилли и Ангельмана – хромосома 15(q11.2-q13) Синдром Видеманна-Беквита - хромосома 11р15.5 Синдром Сильвера-Рассела
- 24. В музее Прадо в Мадриде есть пара картин придворного художника XVII столетия Хуана Карреньо де Миранда
- 26. Синдром Прадера-Вилли Клинические признаки: ожирение, мышечная гипотония, низкий рост, гипогонадизм, гипогенитализм, умственная отсталость различной степени выраженности.
- 27. Синдром Ангельмана
- 29. ОПРЕДЕЛЕНИЕ МИКРОДЕЛЕЦИЙ ХРОМОСОМЫ 15q11.2 ПРИ СИНДРОМАХ ПРАДЕРА-ВИЛЛИ И АНГЕЛЬМАНА МЕТОДОМ FISH (ДНК-зонд SNRPN).
- 31. Причины, приводящие к СПВ и СА. делеция ОРД мутации в мутации сбалансированные генах- ЦИ транслокации кандидатах
- 32. МОЛЕКУЛЯРНАЯ ОРГАНИЗАЦИЯ РАЙОНА ХРОМОСОМЫ 15(q11-q13)
- 33. Доказательство, что делеция гена SNORD116 может вызывать синдром Прадера-Вилли
- 35. Схема функционирования центра импринтинга при синдромах Прадера-Вилли и Ангельмана ЦИ имеет две основных функции: 1) переключение
- 36. Наследование мутаций центра импринтинга, приводящих к невозможности переключения импринта в герминальных клетках: (а) - СПВ: если
- 37. Анализ аллельного метилирования промоторной области гена SNRPN методом блот-гибридизации с использованием рестриктаз NotI, XbaI и ДНК-зонда
- 38. Анализ аллельного метилирования промоторной области гена SNRPN методом метилспецифической ПЦР. В дорожки с четными номерами нанесены
- 40. Синдром Беквита-Видеманна (11р15) Клинические признаки: макросомия, макроглоссия при рождении, пупочная грыжа, насечки на ушах, гипогликемия, гемигипертрофия,
- 42. Молекулярная организация хромосомного района 11р15.5
- 43. Схема организации и функционирования центра импринтинга 1 и 2 при синдроме Видеманна-Беквита
- 44. Структурно-функциональная организация ЦИ1 и ЦИ2. Энхансеры (зеленые овалы) стимулируют транскрипцию днРНК H19 и внутригенной микроРНК miR-675
- 45. Гено-фенотипические корреляции при СВБ 10-15% 7% 55% 20% 2%
- 46. Молекулярная диагностика аллельного метилирования IGF2 при СВБ Сравнение данных денситометрии дорожек 2 и 5
- 47. Однородительская дисомия при СВБ
- 48. Пренатальная и постнатальная задержка роста; Треугольное лицо с выступающим лбом; Клинодактилия или брахидактилия; Макроцефалия; Скелетная асимметрия;
- 49. 7p11.2-p13. У человека отцовская экспрессия GRB10 установлена в головном и спинном мозге, материнская – в скелетных
- 50. Схема эпигенетической патологии при СРС (гипометилирование H19 на отцовской хромосоме 11)
- 51. Схема молекулярной диагностики СРС и СВБ
- 52. Молекулярная диагностика аномального метилирования при СРС и СВБ
- 53. Псевдогипопаратиреоз 1в низкий рост; круглое лицо; задержка нервно-психического развития; скелетные аномалии; низкое содержание кальция в сыворотке
- 54. Псевдогипопаратиреоидизм 1в проявляется гипокальцемией и гиперфосфатемией в результате резистентности к ПТГ. Описаны как спорадические, так и
- 55. Локус GNAS1 имеет три альтернативных первых экзона (А/B, XL и NESP55), которые сплайсируются с 2-13 экзонами,
- 56. Структурная молекулярная патология, которая приводит к нарушению функционирования импринтированного локуса GNAS. Делеция 3 т.п.н. гена STX16
- 57. Район хромосомы 14q32.2 содержит кластер импринтированных генов: часть экспрессируется с отцовской хромосомы – DLK1, RTL1 и
- 58. Импринтированный район хромосомы 14q32.2 и экспрессия импринтированных генов. Гены, имеющие отцовскую экспрессию, обозначены синим цветом, материнскую
- 59. Некоторые миРНК млекопитающих импринтированы. У мыши miR-127 и miR-136 транскрибируются как анти-смысловые к реципрокно импринтированному транспозон-подобному
- 61. Схема молекулярной организации импринтированного района 6q24. Транзиторный неонатальный диабет редкое заболевание (частота 1:500000 новорожденных), которое проявляется
- 62. Структура импринтированного района 6q24.2 и варианты экспрессии генов при ТНСД Розовые прямоугольники и стрелки – биаллельно
- 63. Описано более 10 пациентов с СВБ, у которых, помимо материнского гипометилирования СВБ-ЦИ2 обнаружена потеря метилирования по
- 64. Импринтинг и вспомогательные репродуктивные технологии Наиболее распространенная патология: С-м Ангельмана – 20 случаев С-м- Прадера-Вилли –
- 65. Целый ряд причин может приводить к эпигенетическим аномалиям: 1) бесплодие само по себе; 2) процесс стимуляции
- 66. Метилирование/деметилирование в процессе гаметогенеза
- 67. Критические этапы гаметогенеза и раннего эмбриогенеза, могущие привести к эпигенетической патологии при ВРТ
- 68. Синдром Мартина-Белл
- 69. Синдром Мартина-Белл
- 70. Наследование СМБ носит необычный характер: передача заболевания происходит через фенотипически нормальных мужчин (нормальные трансмиттеры); дочери нормальных
- 71. Ломкость Х-хромосомы
- 72. Схема расположения фолатчувствительных ломких сайтов на Х-хромосоме
- 73. Синдром Мартина-Белл Аллели в состоянии премутации выявляются у всех нормальных трансмиттеров и, по крайней мере, у
- 76. Гипотетическая родословная и результаты блот-гибридизации по Саузерну, иллюстрирующие применение системы EcoRI+EagI/StB12.3 в диагностике СМБ. 1, 7
- 77. ДНК-диагностика синдрома Мартина-Белл Анализ ДНК здоровых и больных индивидов, обработанной рестриктазами EcoRI и EagI, методом гибридизации
- 78. Детекция длин CGG-повторов в первом экзоне FMR1 методом МС-ПЦР с флуоресцентно меченым праймером. Продукты ПЦР разделены
- 79. ПЦР-тест на метилирование промоторной области гена FMR1 (МЧ-ПЦР). 1 – Маркёр молекулярной массы. 2,3 – ДНК
- 80. Анализ метилирования и длин CGG-повтора гена FMR1 у пациентов женского пола методом метилспецифической ПЦР. Образец 1
- 81. Анализ метилирования CpG-островков FRAXA, FRAXE и FRAXF 1 2 3 4 5 6 7 8 9
- 82. Алгоритм неонатального скрининга мальчиков на основе анализа метилирования CpG-островков FRAXA, FRAXE и FRAXF.
- 83. Предполагается, что геном человека содержит 100 – 200 импринтированных генов. На сегодняшний день их около 100
- 84. Характерные черты импринтированных генов 1. Кластеризация. Импринтированные гены распределены не случайным образом в геноме, а встречаются
- 85. 2. Консервативность импринтинга. Характер импринтинга генов H19, IGF2, p57KIP и SNRPN идентичен у человека и мыши.
- 86. 4. Онтогенетическая и тканевая регуляция импринтинга. INS2 импринтирован только в экстраэмбриональных тканях мышиного эмбриона, но экспрессируется
- 87. Практически все импринтированные гены содержат повторы, в частности, первый интрон гена SNRPN содержит структурно консервативные G-обогащенные
- 88. 5. Импринтированные гены кодируют как белки, так и только РНК. Некоторые импринтированные гены не кодируют белков,
- 89. Некодирующие РНК импринтированных районов
- 90. Общая модель организации импринтированного района. IC – центр импринтинга; IME – элемент, поддерживающий импринтинг. Центр импринтинга
- 91. ХАРАКТЕРНЫЕ ЧЕРТЫ ЦЕНТРОВ ИМПРИНТИНГА 1. Регулируют импринтированные гены в кластере in cis; 2. Имеют дифференциальное аллельное
- 93. Доменная организация хроматина в ядре
- 94. Подвижность генов – важный фактор их регуляции. Кластеры активных генов - 150-200 т.п.н., расстояние между кластерами
- 95. ПЕТЛЕВАЯ МОДЕЛЬ РЕГУЛЯЦИИ ИМПРИНТИРОВАННОГО РАЙОНА Центры импринтинга выполняют функцию перемещения двух дифференциально метилированных аллелей в различные
- 101. ПЕТЛЕВАЯ МОДЕЛЬ РЕГУЛЯЦИИ ИМПРИНТИРОВАННОГО РАЙОНА
- 107. Однородительская дисомия и ранняя эмбриолетальность
- 108. Целый ряд заболеваний по характеру наследования и проявлениям может возникать вследствие импринтинга. Синдром Вильямса с тяжелыми
- 109. Схема регуляции экспрессии генов при лице-плече-лопаточной мышечной дистрофии. Гиперэкспрессия гена ANT1 индуцирует апоптоз, характерный для мышечных
- 110. Синдром ломкой Х хромосомы (возможный сценарий) CGG повтор в 5’ НТР гена FMR1 в результате экспансии
- 111. Метилирование ДНК посредством siРНК
- 113. Скачать презентацию

Геномный импринтинг - эпигенетический механизм регуляции экспрессии гомологичных генов в
Геномный импринтинг - эпигенетический механизм регуляции экспрессии гомологичных генов в
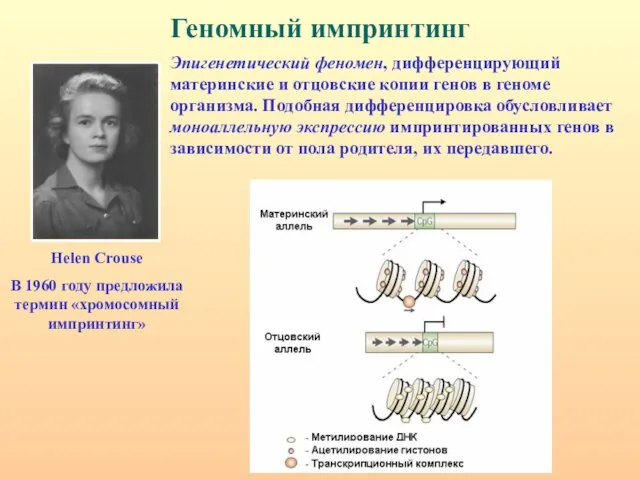
Геномный импринтинг
Эпигенетический феномен, дифференцирующий материнские и отцовские копии генов в геноме
Геномный импринтинг
Эпигенетический феномен, дифференцирующий материнские и отцовские копии генов в геноме
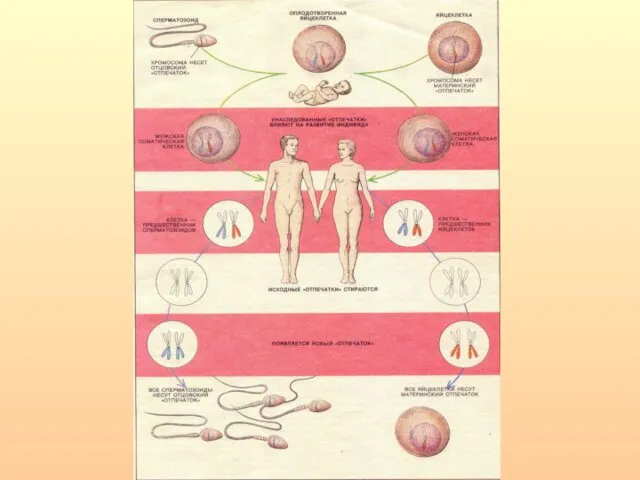
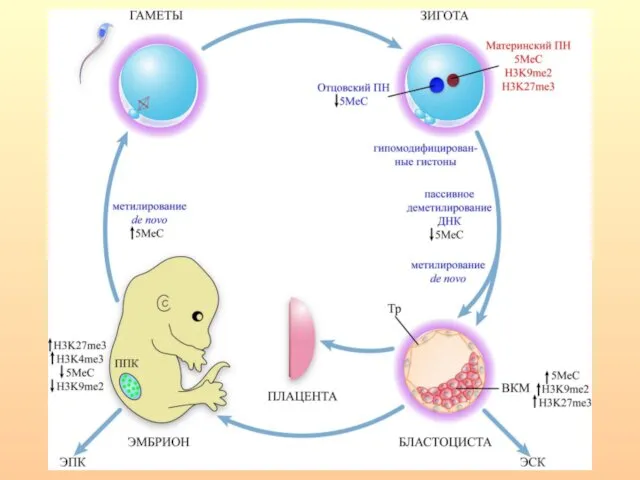
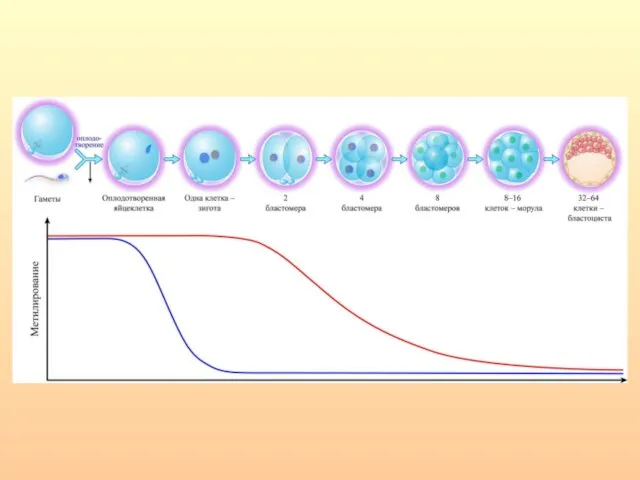
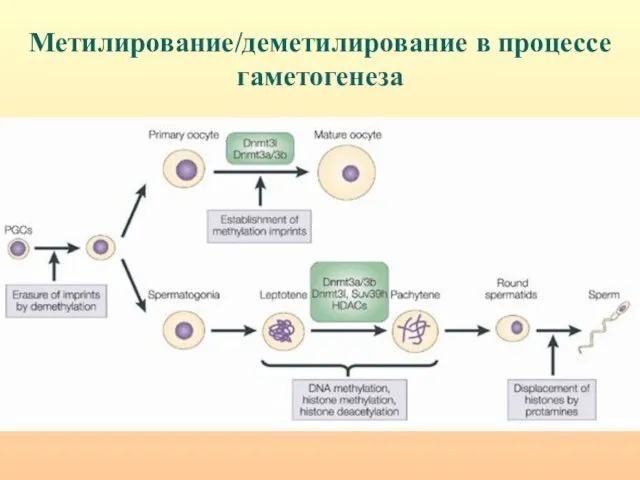
Метилирование/деметилирование в процессе гаметогенеза
Метилирование/деметилирование в процессе гаметогенеза
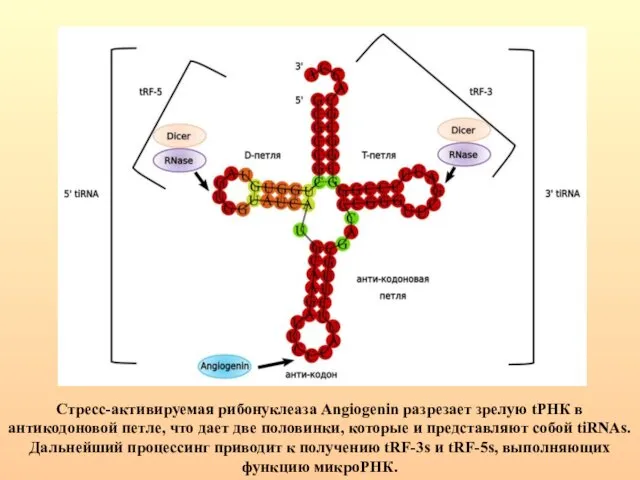
Стресс-активируемая рибонуклеаза Angiogenin разрезает зрелую tРНК в антикодоновой петле, что дает
Стресс-активируемая рибонуклеаза Angiogenin разрезает зрелую tРНК в антикодоновой петле, что дает

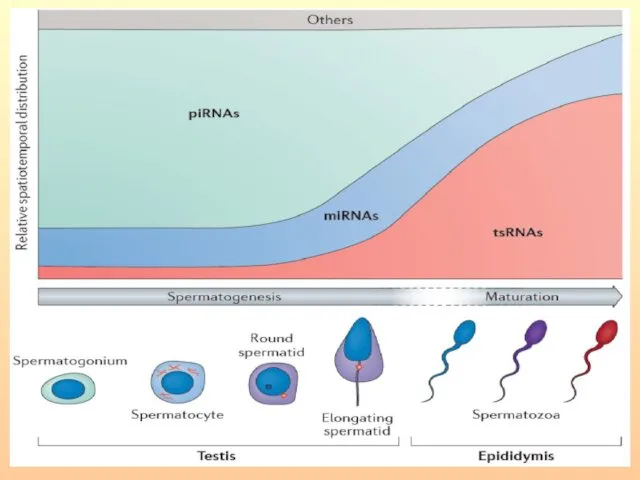
- Несколько сотен важных для развития малых РНК соматического происхождения
- Несколько сотен важных для развития малых РНК соматического происхождения
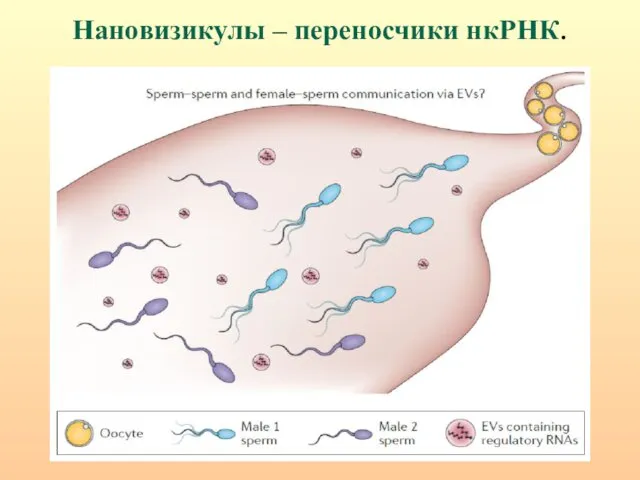
Нановизикулы – переносчики нкРНК.
Нановизикулы – переносчики нкРНК.

Барьер Вейсмана – миф!
Tелегония на марше:
(проявление признаков первого самца у потомства
Барьер Вейсмана – миф! Tелегония на марше: (проявление признаков первого самца у потомства

Tелегония
В XIX в. лорд Мортон, близкий друг Ч. Дарвина отважился
Tелегония
В XIX в. лорд Мортон, близкий друг Ч. Дарвина отважился

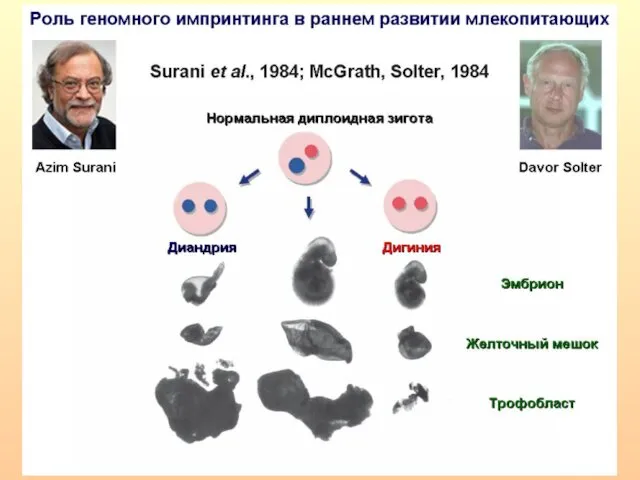
Для нормального развития необходим равный вклад обоих родителей.
Трансплантация пронуклеусов.
Андрогенетические зиготы
Для нормального развития необходим равный вклад обоих родителей.
Трансплантация пронуклеусов.
Андрогенетические зиготы

Наши основоположники
В.С. Баранов
А.П. Дыбан, 1922-2002
А.П. Дыбан и В.С.Баранов внесли значительный
Наши основоположники
В.С. Баранов
А.П. Дыбан, 1922-2002
А.П. Дыбан и В.С.Баранов внесли значительный

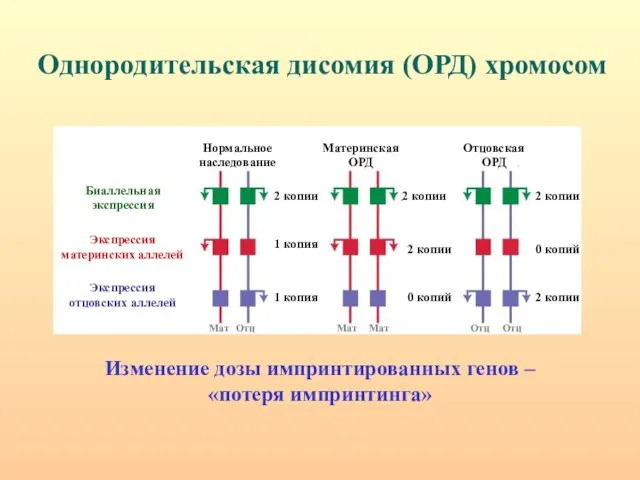
Однородительская дисомия.
На мышиных транслокационных гибридах, несущих отдельные хромосомные участки, имеющие как
Однородительская дисомия.
На мышиных транслокационных гибридах, несущих отдельные хромосомные участки, имеющие как

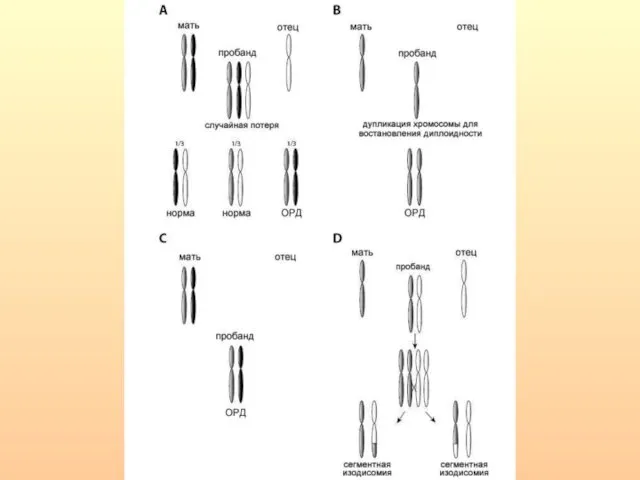
Механизмы формирования ОРД у человека.
1) Комплементация гамет – дополнение
Механизмы формирования ОРД у человека.
1) Комплементация гамет – дополнение

ОРД по целым хромосомам или их фрагментам выявлены при анализе наследственной
ОРД по целым хромосомам или их фрагментам выявлены при анализе наследственной
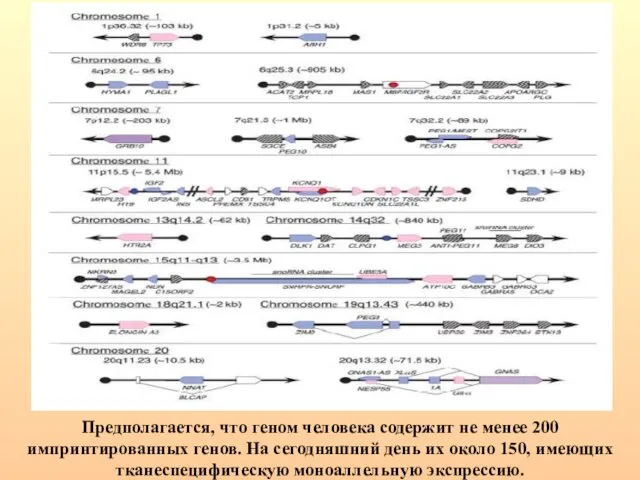
Предполагается, что геном человека содержит не менее 200
импринтированных генов. На
Предполагается, что геном человека содержит не менее 200
импринтированных генов. На

Болезни импринтинга
Синдромы Прадера-Вилли и Ангельмана –
хромосома 15(q11.2-q13)
Синдром Видеманна-Беквита - хромосома
Болезни импринтинга
Синдромы Прадера-Вилли и Ангельмана –
хромосома 15(q11.2-q13)
Синдром Видеманна-Беквита - хромосома

В музее Прадо в Мадриде есть пара картин придворного художника XVII
В музее Прадо в Мадриде есть пара картин придворного художника XVII
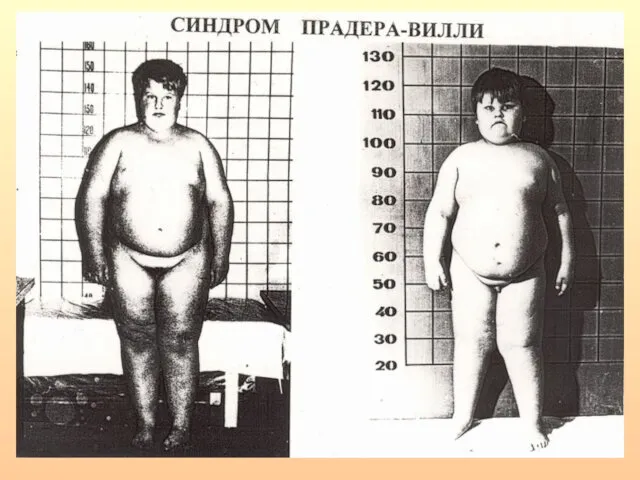
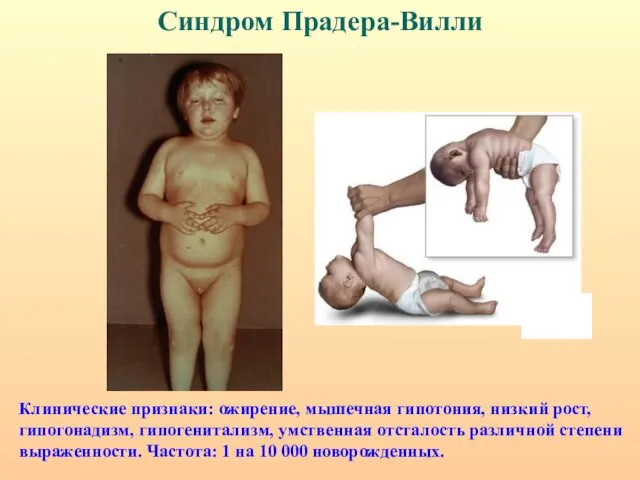
Синдром Прадера-Вилли
Клинические признаки: ожирение, мышечная гипотония, низкий рост, гипогонадизм, гипогенитализм,
Синдром Прадера-Вилли
Клинические признаки: ожирение, мышечная гипотония, низкий рост, гипогонадизм, гипогенитализм,

Синдром Ангельмана
Синдром Ангельмана

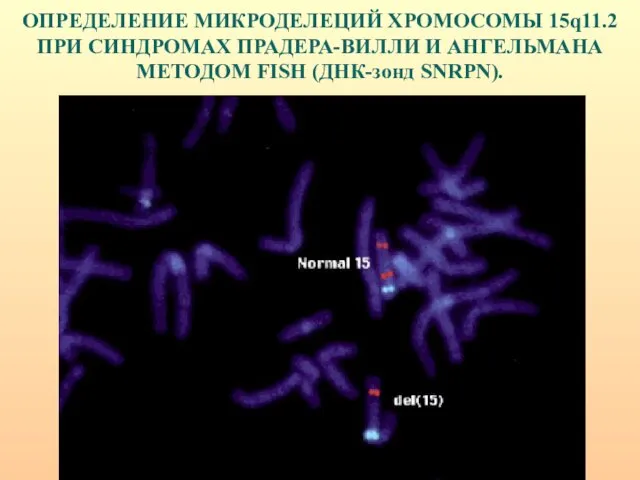
ОПРЕДЕЛЕНИЕ МИКРОДЕЛЕЦИЙ ХРОМОСОМЫ 15q11.2 ПРИ СИНДРОМАХ ПРАДЕРА-ВИЛЛИ И АНГЕЛЬМАНА МЕТОДОМ FISH
ОПРЕДЕЛЕНИЕ МИКРОДЕЛЕЦИЙ ХРОМОСОМЫ 15q11.2 ПРИ СИНДРОМАХ ПРАДЕРА-ВИЛЛИ И АНГЕЛЬМАНА МЕТОДОМ FISH
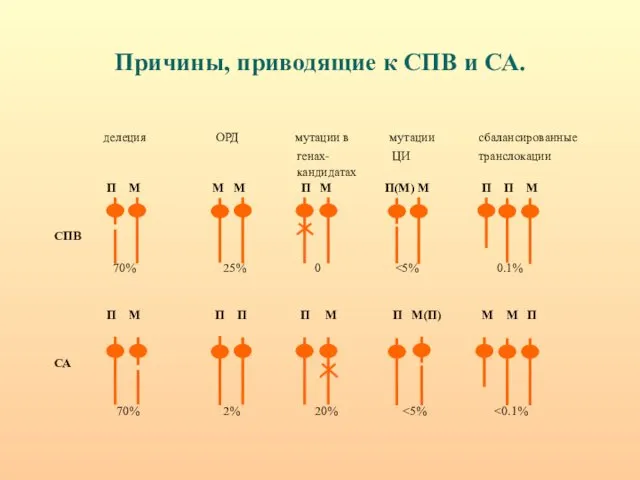
Причины, приводящие к СПВ и СА.
делеция ОРД мутации в мутации
Причины, приводящие к СПВ и СА.
делеция ОРД мутации в мутации
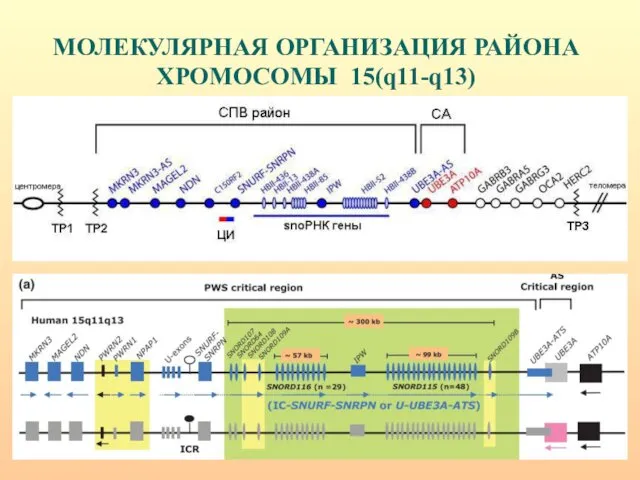
МОЛЕКУЛЯРНАЯ ОРГАНИЗАЦИЯ РАЙОНА
ХРОМОСОМЫ 15(q11-q13)
МОЛЕКУЛЯРНАЯ ОРГАНИЗАЦИЯ РАЙОНА
ХРОМОСОМЫ 15(q11-q13)
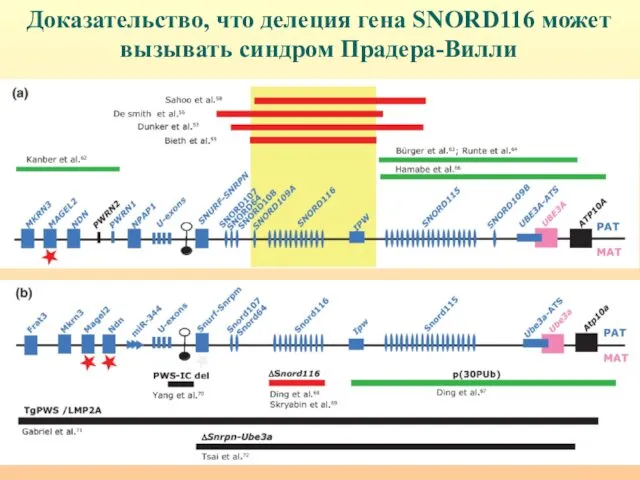
Доказательство, что делеция гена SNORD116 может вызывать синдром Прадера-Вилли
Доказательство, что делеция гена SNORD116 может вызывать синдром Прадера-Вилли
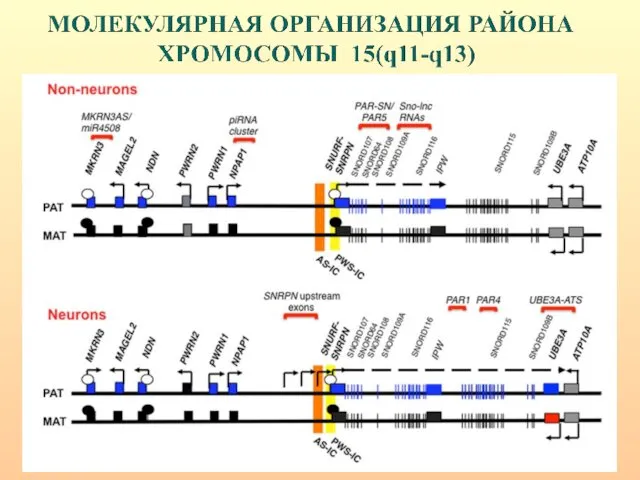
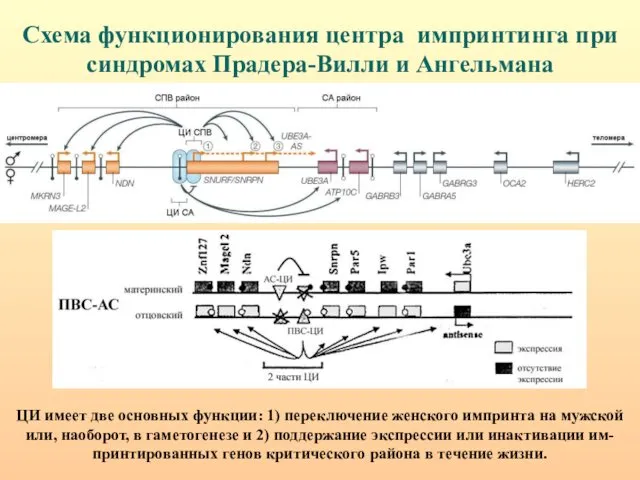
Схема функционирования центра импринтинга при синдромах Прадера-Вилли и Ангельмана
ЦИ имеет две
Схема функционирования центра импринтинга при синдромах Прадера-Вилли и Ангельмана
ЦИ имеет две
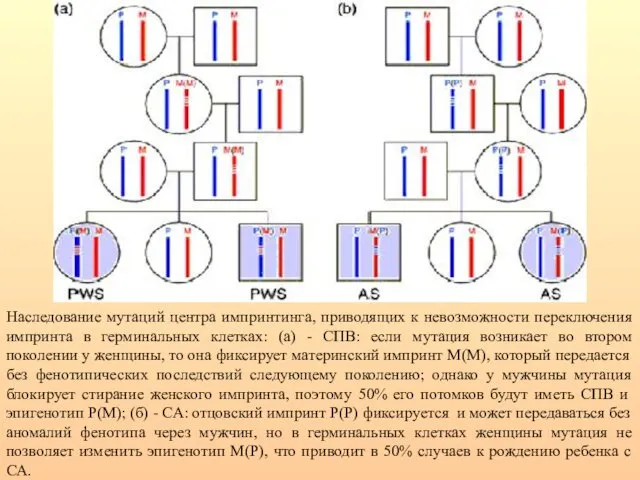
Наследование мутаций центра импринтинга, приводящих к невозможности переключения импринта в герминальных
Наследование мутаций центра импринтинга, приводящих к невозможности переключения импринта в герминальных
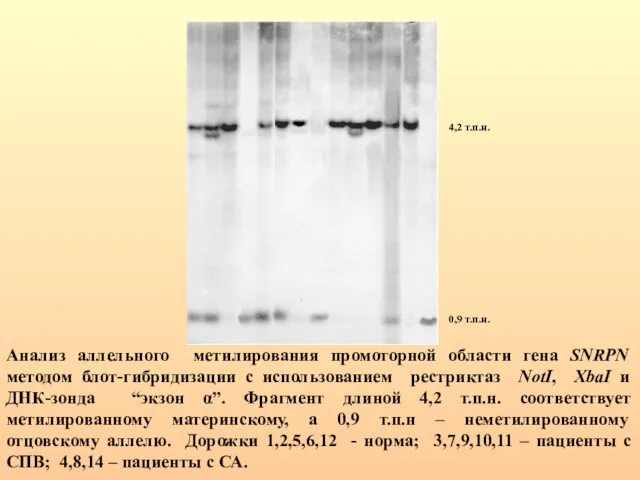
Анализ аллельного метилирования промоторной области гена SNRPN методом блот-гибридизации с использованием
Анализ аллельного метилирования промоторной области гена SNRPN методом блот-гибридизации с использованием
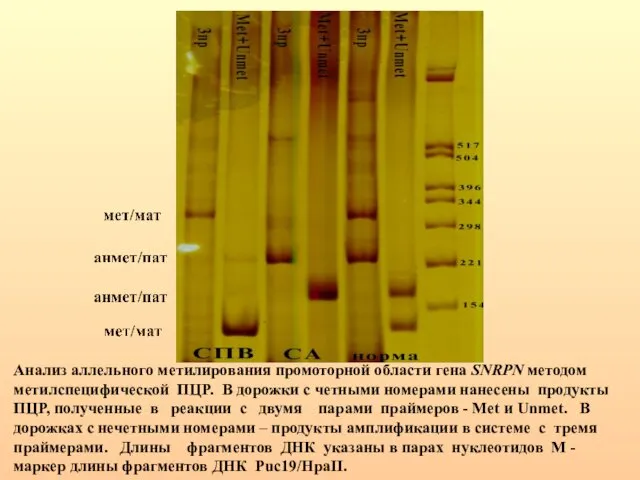
Анализ аллельного метилирования промоторной области гена SNRPN методом метилспецифической ПЦР. В
Анализ аллельного метилирования промоторной области гена SNRPN методом метилспецифической ПЦР. В
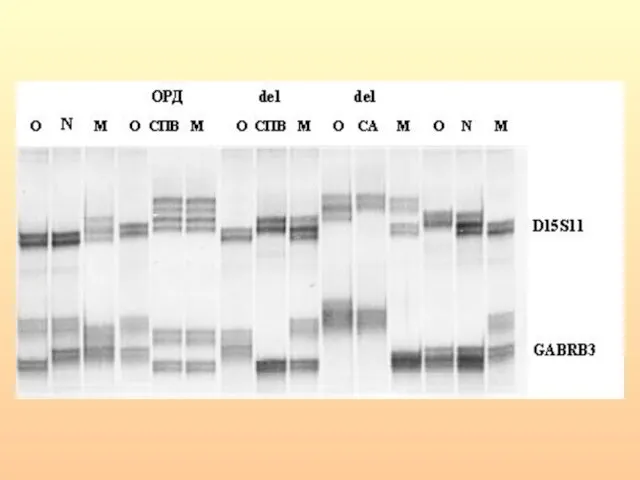
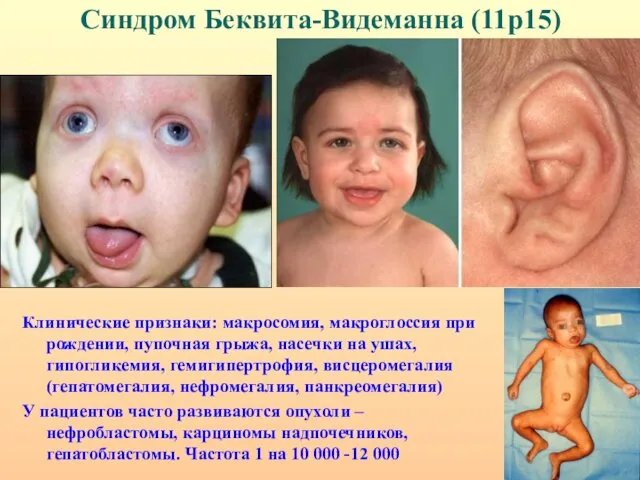
Синдром Беквита-Видеманна (11р15)
Клинические признаки: макросомия, макроглоссия при рождении, пупочная грыжа, насечки
Синдром Беквита-Видеманна (11р15)
Клинические признаки: макросомия, макроглоссия при рождении, пупочная грыжа, насечки
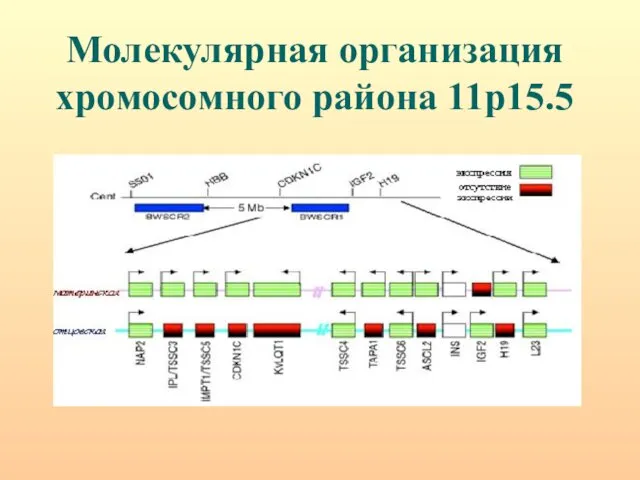
Молекулярная организация хромосомного района 11р15.5
Молекулярная организация хромосомного района 11р15.5
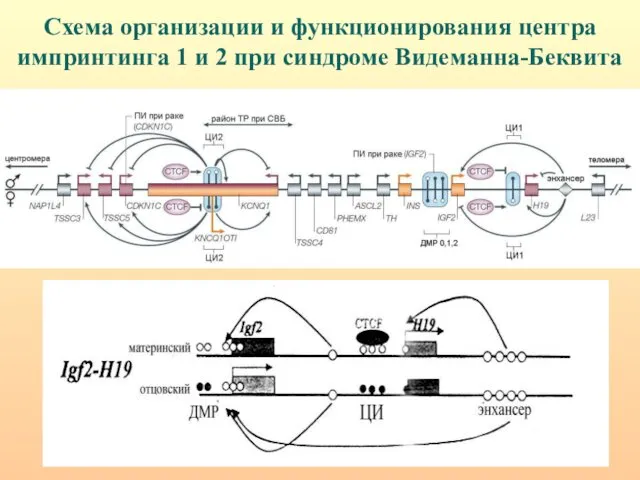
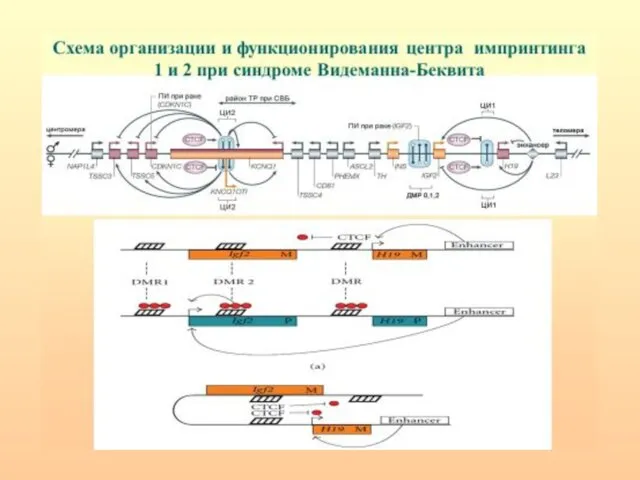
Схема организации и функционирования центра импринтинга 1 и 2 при синдроме
Схема организации и функционирования центра импринтинга 1 и 2 при синдроме
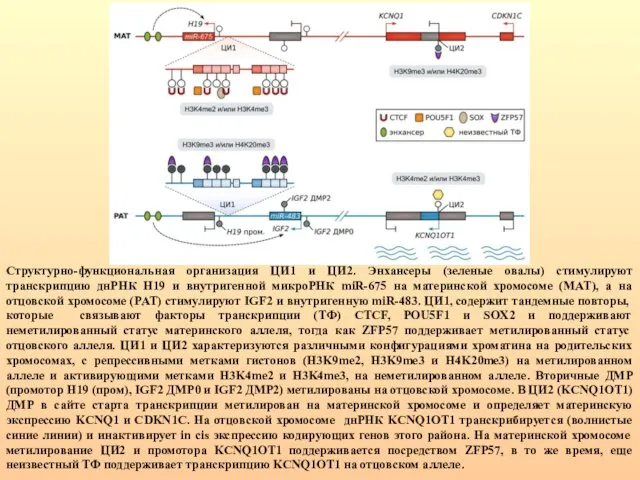
Структурно-функциональная организация ЦИ1 и ЦИ2. Энхансеры (зеленые овалы) стимулируют транскрипцию днРНК
Структурно-функциональная организация ЦИ1 и ЦИ2. Энхансеры (зеленые овалы) стимулируют транскрипцию днРНК

Гено-фенотипические корреляции при СВБ
10-15%
7%
55%
20%
2%
Гено-фенотипические корреляции при СВБ
10-15%
7%
55%
20%
2%
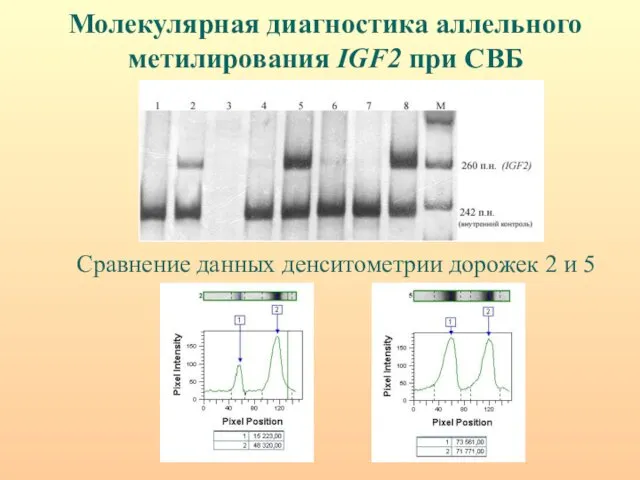
Молекулярная диагностика аллельного метилирования IGF2 при СВБ
Сравнение данных денситометрии дорожек 2
Молекулярная диагностика аллельного метилирования IGF2 при СВБ
Сравнение данных денситометрии дорожек 2

Однородительская дисомия при СВБ
Однородительская дисомия при СВБ

Пренатальная и постнатальная задержка роста;
Треугольное лицо с выступающим лбом;
Клинодактилия или брахидактилия;
Макроцефалия;
Скелетная
Пренатальная и постнатальная задержка роста;
Треугольное лицо с выступающим лбом;
Клинодактилия или брахидактилия;
Макроцефалия;
Скелетная

7p11.2-p13. У человека отцовская экспрессия GRB10 установлена в головном и
7p11.2-p13. У человека отцовская экспрессия GRB10 установлена в головном и
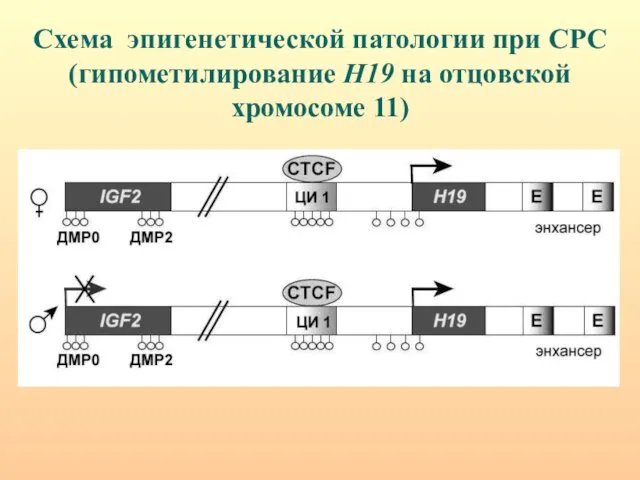
Схема эпигенетической патологии при СРС
(гипометилирование H19 на отцовской хромосоме 11)
Схема эпигенетической патологии при СРС
(гипометилирование H19 на отцовской хромосоме 11)
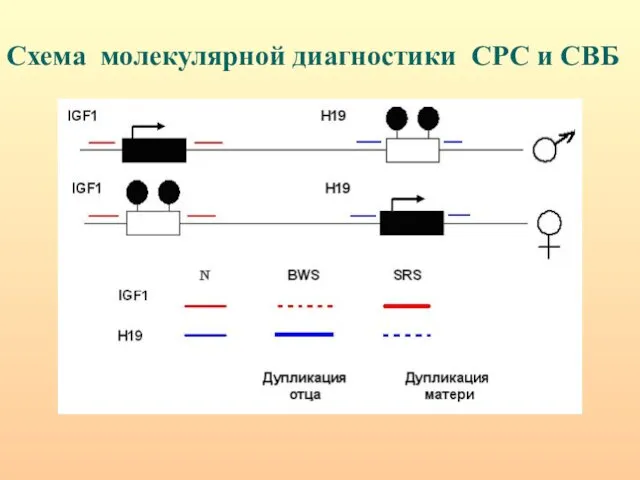
Схема молекулярной диагностики СРС и СВБ
Схема молекулярной диагностики СРС и СВБ
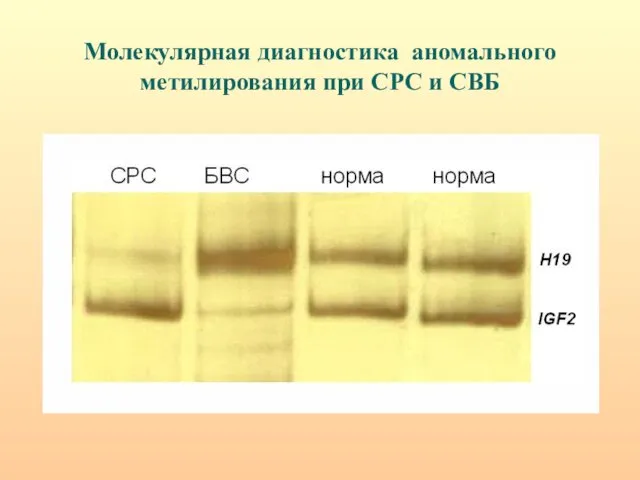
Молекулярная диагностика аномального метилирования при СРС и СВБ
Молекулярная диагностика аномального метилирования при СРС и СВБ
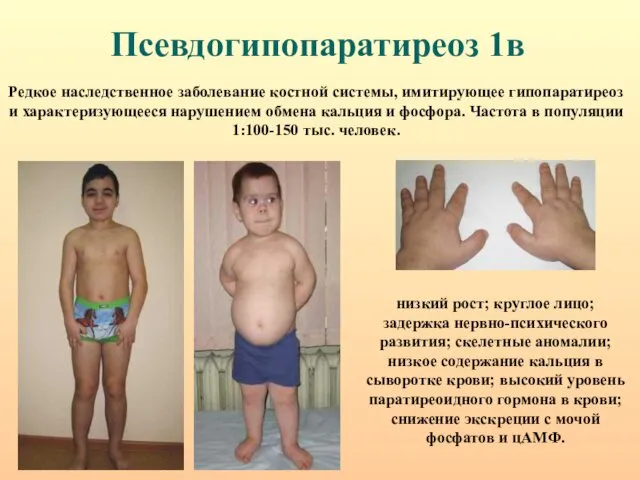
Псевдогипопаратиреоз 1в
низкий рост; круглое лицо;
задержка нервно-психического развития; скелетные аномалии;
низкое содержание
Псевдогипопаратиреоз 1в
низкий рост; круглое лицо;
задержка нервно-психического развития; скелетные аномалии;
низкое содержание

Псевдогипопаратиреоидизм 1в проявляется гипокальцемией и гиперфосфатемией в результате резистентности к ПТГ.
Псевдогипопаратиреоидизм 1в проявляется гипокальцемией и гиперфосфатемией в результате резистентности к ПТГ.

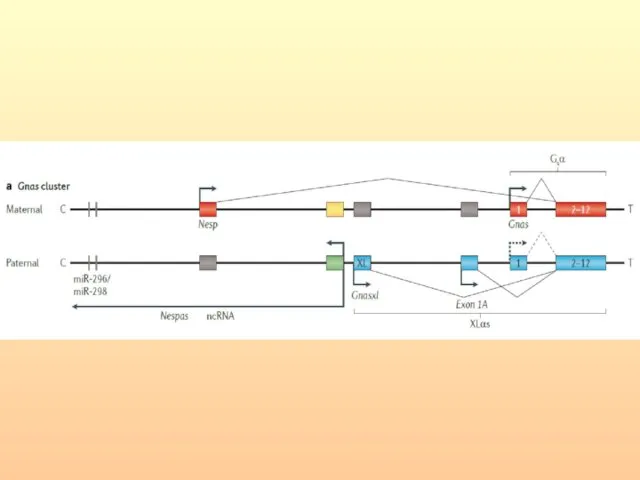
Локус GNAS1 имеет три альтернативных первых экзона (А/B, XL и NESP55),
Локус GNAS1 имеет три альтернативных первых экзона (А/B, XL и NESP55),
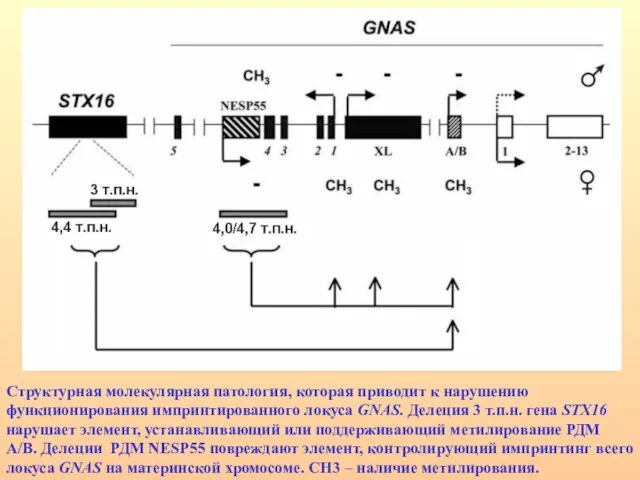
Структурная молекулярная патология, которая приводит к нарушению функционирования импринтированного локуса GNAS.
Структурная молекулярная патология, которая приводит к нарушению функционирования импринтированного локуса GNAS.

Район хромосомы 14q32.2 содержит кластер импринтированных генов: часть экспрессируется с отцовской
Район хромосомы 14q32.2 содержит кластер импринтированных генов: часть экспрессируется с отцовской
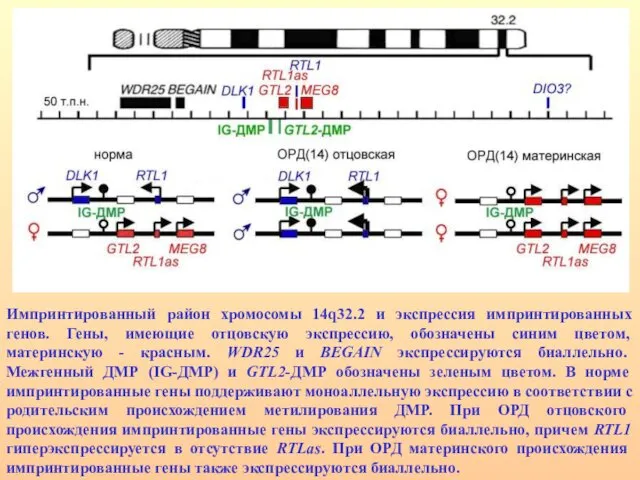
Импринтированный район хромосомы 14q32.2 и экспрессия импринтированных генов. Гены, имеющие отцовскую
Импринтированный район хромосомы 14q32.2 и экспрессия импринтированных генов. Гены, имеющие отцовскую

Некоторые миРНК млекопитающих импринтированы. У мыши miR-127 и miR-136 транскрибируются
Некоторые миРНК млекопитающих импринтированы. У мыши miR-127 и miR-136 транскрибируются
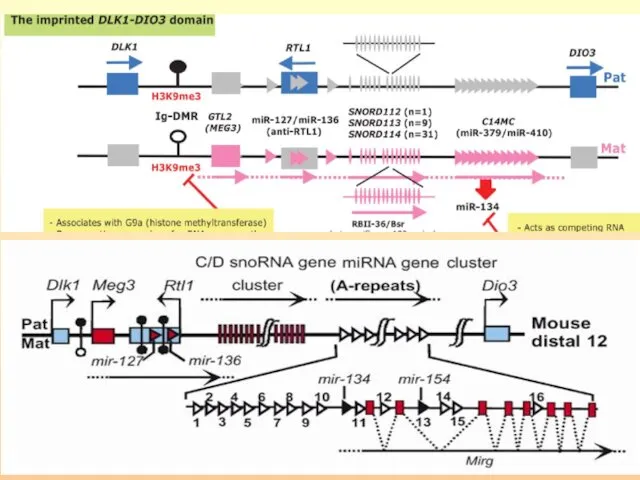

Схема молекулярной организации импринтированного района 6q24.
Транзиторный неонатальный диабет редкое заболевание
Схема молекулярной организации импринтированного района 6q24.
Транзиторный неонатальный диабет редкое заболевание
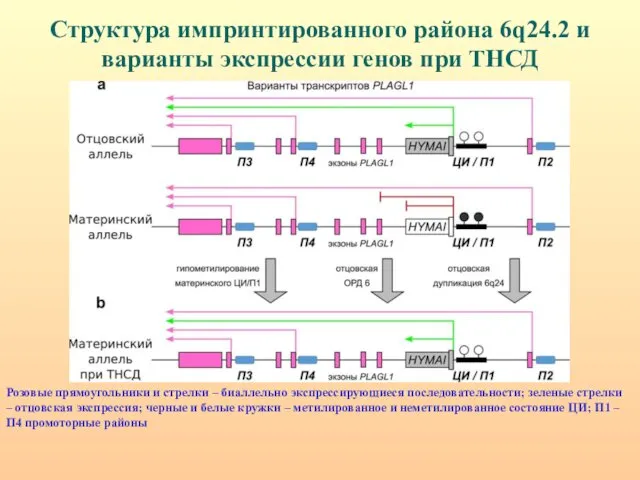
Структура импринтированного района 6q24.2 и варианты экспрессии генов при ТНСД
Розовые
Структура импринтированного района 6q24.2 и варианты экспрессии генов при ТНСД
Розовые

Описано более 10 пациентов с СВБ, у которых, помимо материнского гипометилирования
Описано более 10 пациентов с СВБ, у которых, помимо материнского гипометилирования

Импринтинг
и вспомогательные репродуктивные технологии
Наиболее распространенная патология:
С-м Ангельмана – 20 случаев
С-м-
Импринтинг
и вспомогательные репродуктивные технологии
Наиболее распространенная патология:
С-м Ангельмана – 20 случаев
С-м-

Целый ряд причин может приводить к эпигенетическим аномалиям: 1) бесплодие само
Целый ряд причин может приводить к эпигенетическим аномалиям: 1) бесплодие само
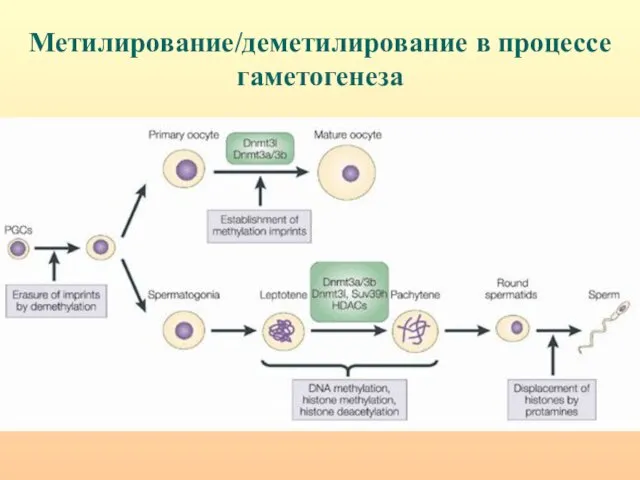
Метилирование/деметилирование в процессе гаметогенеза
Метилирование/деметилирование в процессе гаметогенеза
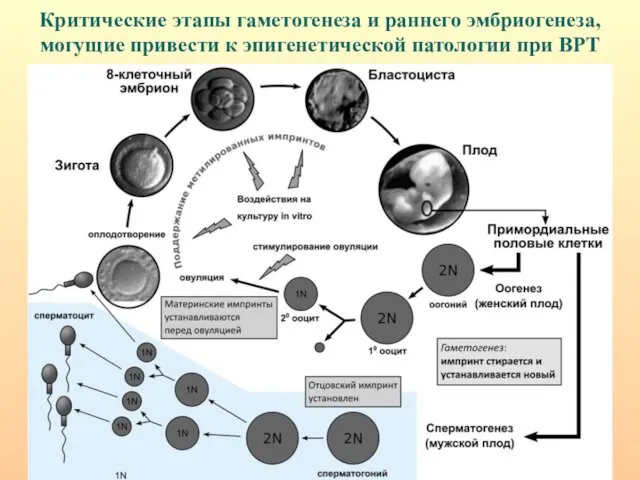
Критические этапы гаметогенеза и раннего эмбриогенеза, могущие привести к эпигенетической патологии
Критические этапы гаметогенеза и раннего эмбриогенеза, могущие привести к эпигенетической патологии

Синдром Мартина-Белл
Синдром Мартина-Белл
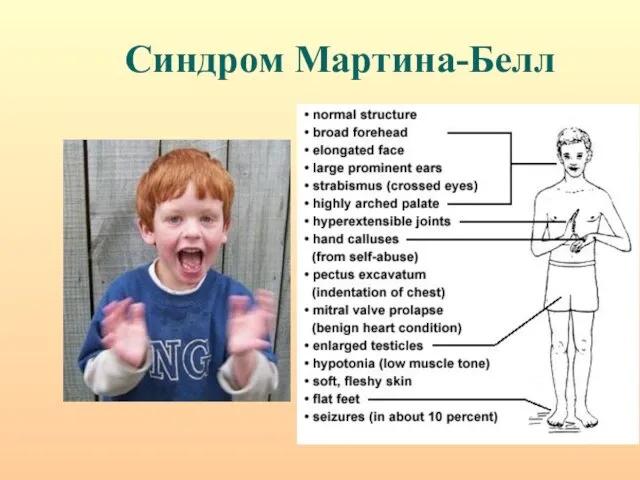
Синдром Мартина-Белл
Синдром Мартина-Белл

Наследование СМБ носит необычный характер:
передача заболевания происходит через фенотипически нормальных
Наследование СМБ носит необычный характер:
передача заболевания происходит через фенотипически нормальных
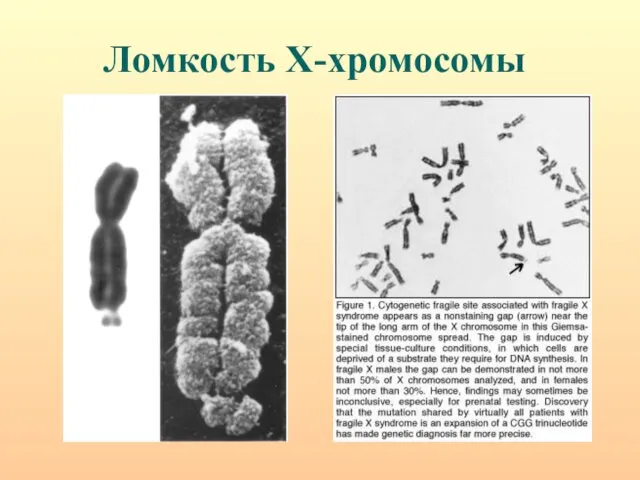
Ломкость Х-хромосомы
Ломкость Х-хромосомы
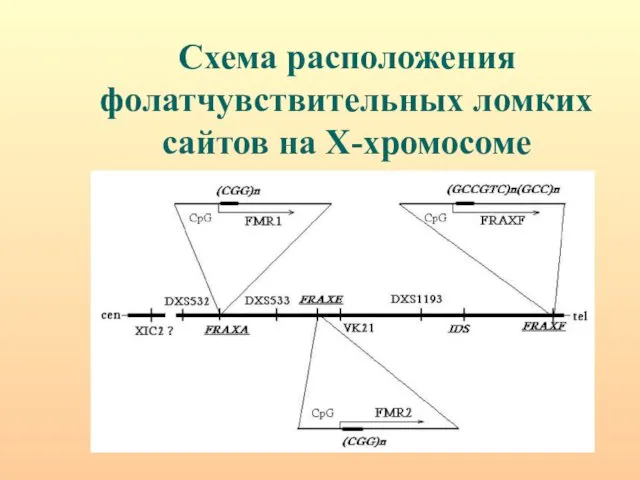
Схема расположения фолатчувствительных ломких сайтов на Х-хромосоме
Схема расположения фолатчувствительных ломких сайтов на Х-хромосоме

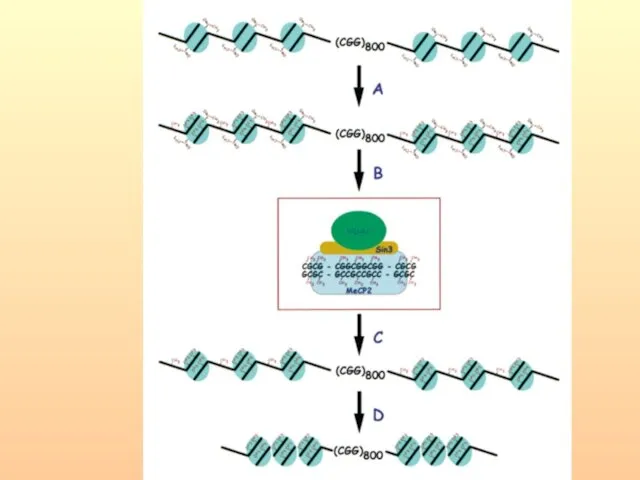
Синдром Мартина-Белл
Аллели в состоянии премутации выявляются у всех нормальных трансмиттеров и,
Синдром Мартина-Белл
Аллели в состоянии премутации выявляются у всех нормальных трансмиттеров и,

Гипотетическая родословная и результаты блот-гибридизации по Саузерну, иллюстрирующие применение системы EcoRI+EagI/StB12.3
Гипотетическая родословная и результаты блот-гибридизации по Саузерну, иллюстрирующие применение системы EcoRI+EagI/StB12.3
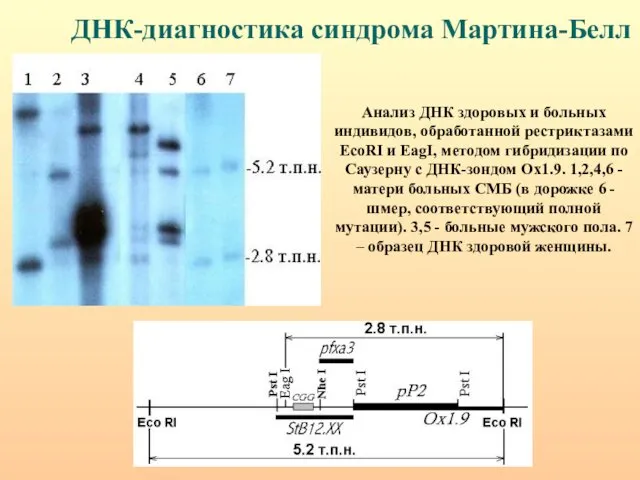
ДНК-диагностика синдрома Мартина-Белл
Анализ ДНК здоровых и больных индивидов, обработанной рестриктазами EcoRI
ДНК-диагностика синдрома Мартина-Белл
Анализ ДНК здоровых и больных индивидов, обработанной рестриктазами EcoRI
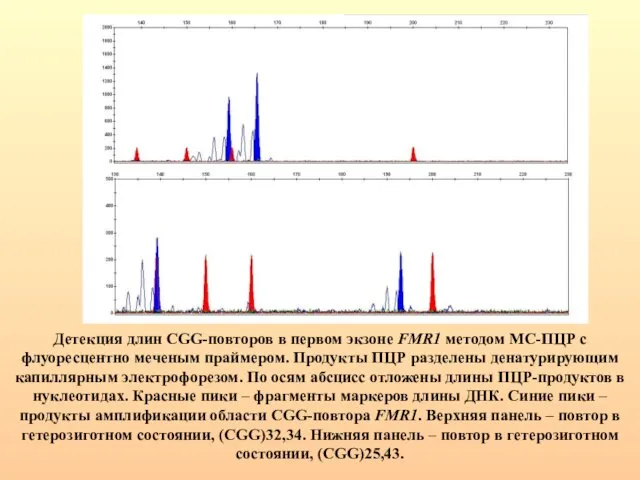
Детекция длин CGG-повторов в первом экзоне FMR1 методом МС-ПЦР с флуоресцентно
Детекция длин CGG-повторов в первом экзоне FMR1 методом МС-ПЦР с флуоресцентно
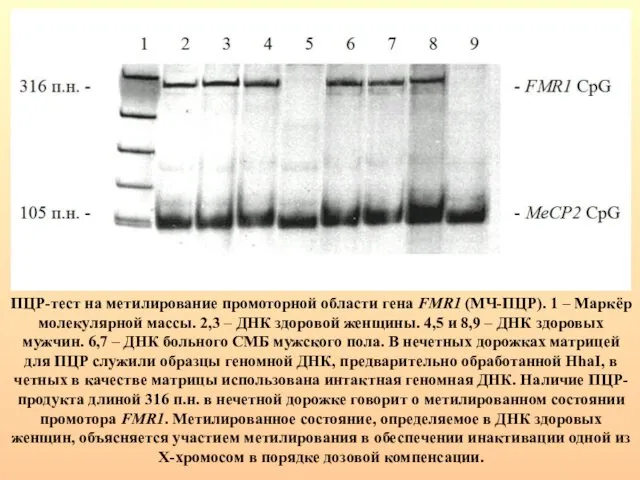
ПЦР-тест на метилирование промоторной области гена FMR1 (МЧ-ПЦР). 1 – Маркёр
ПЦР-тест на метилирование промоторной области гена FMR1 (МЧ-ПЦР). 1 – Маркёр
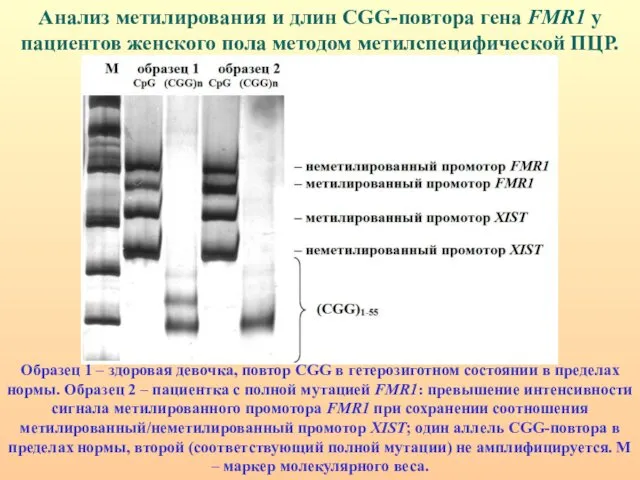
Анализ метилирования и длин CGG-повтора гена FMR1 у пациентов женского пола
Анализ метилирования и длин CGG-повтора гена FMR1 у пациентов женского пола
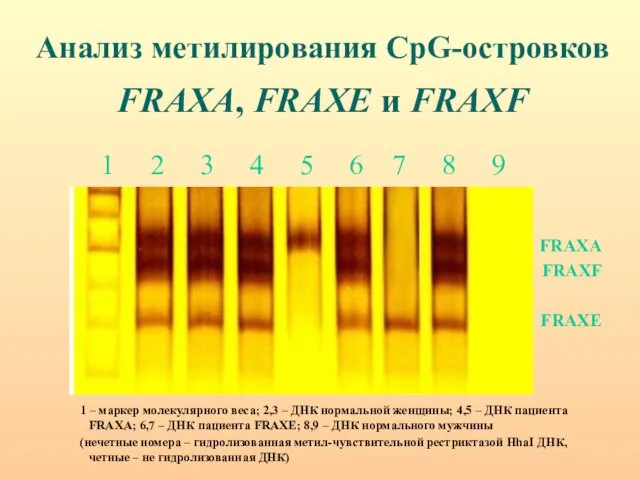
Анализ метилирования CpG-островков
FRAXA, FRAXE и FRAXF
1 2 3
Анализ метилирования CpG-островков
FRAXA, FRAXE и FRAXF
1 2 3
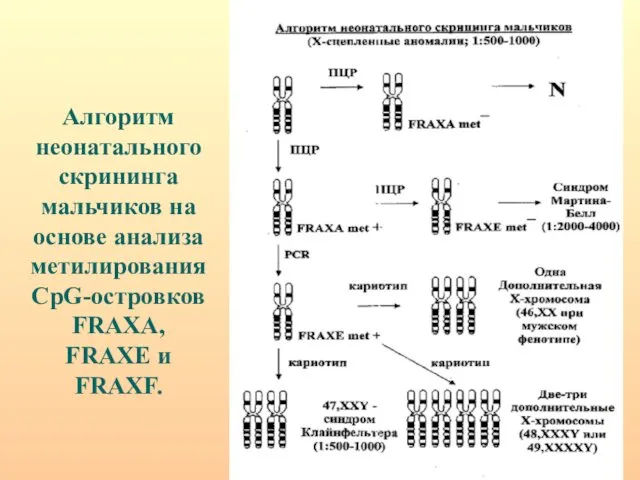
Алгоритм неонатального скрининга мальчиков на основе анализа метилирования CpG-островков FRAXA, FRAXE
Алгоритм неонатального скрининга мальчиков на основе анализа метилирования CpG-островков FRAXA, FRAXE
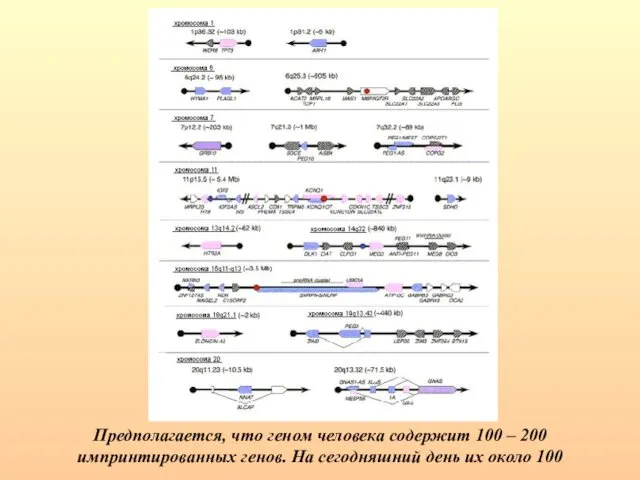
Предполагается, что геном человека содержит 100 – 200
импринтированных генов. На
Предполагается, что геном человека содержит 100 – 200
импринтированных генов. На

Характерные черты импринтированных генов
1. Кластеризация.
Импринтированные гены распределены не случайным образом в
Характерные черты импринтированных генов
1. Кластеризация.
Импринтированные гены распределены не случайным образом в

2. Консервативность импринтинга.
Характер импринтинга генов H19, IGF2, p57KIP и SNRPN
2. Консервативность импринтинга.
Характер импринтинга генов H19, IGF2, p57KIP и SNRPN

4. Онтогенетическая и тканевая регуляция
импринтинга.
INS2 импринтирован только в экстраэмбриональных тканях мышиного
4. Онтогенетическая и тканевая регуляция
импринтинга.
INS2 импринтирован только в экстраэмбриональных тканях мышиного

Практически все импринтированные гены содержат повторы, в частности, первый интрон гена
Практически все импринтированные гены содержат повторы, в частности, первый интрон гена

5. Импринтированные гены кодируют как белки, так и только РНК.
Некоторые импринтированные
5. Импринтированные гены кодируют как белки, так и только РНК.
Некоторые импринтированные
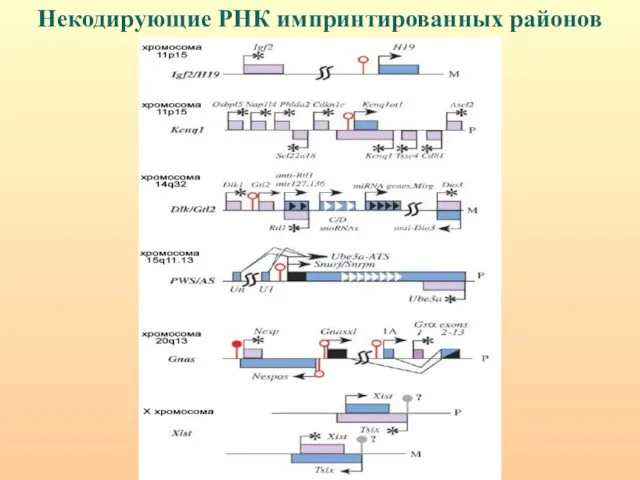
Некодирующие РНК импринтированных районов
Некодирующие РНК импринтированных районов
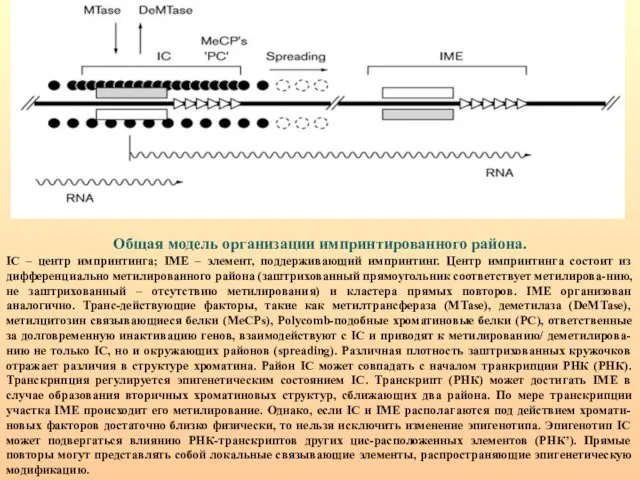
Общая модель организации импринтированного района.
IC – центр импринтинга; IME – элемент,
Общая модель организации импринтированного района.
IC – центр импринтинга; IME – элемент,
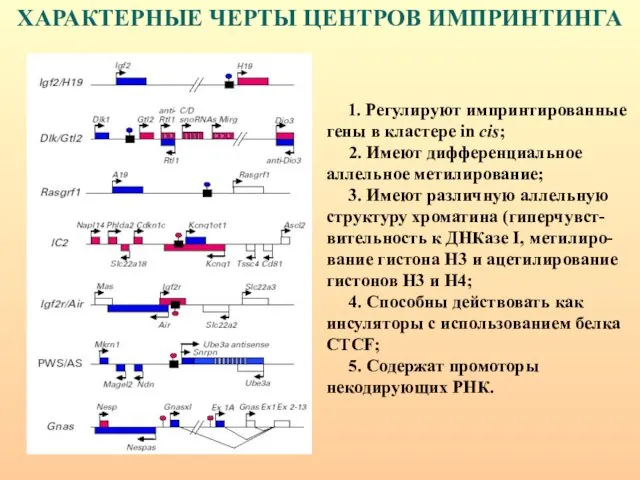
ХАРАКТЕРНЫЕ ЧЕРТЫ ЦЕНТРОВ ИМПРИНТИНГА
1. Регулируют импринтированные гены в кластере in
ХАРАКТЕРНЫЕ ЧЕРТЫ ЦЕНТРОВ ИМПРИНТИНГА
1. Регулируют импринтированные гены в кластере in
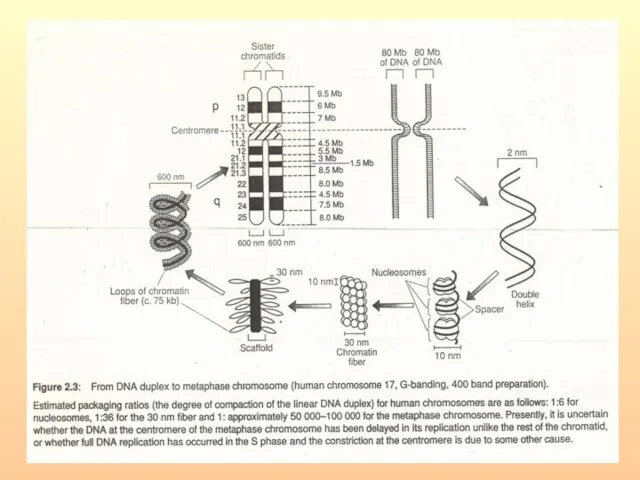
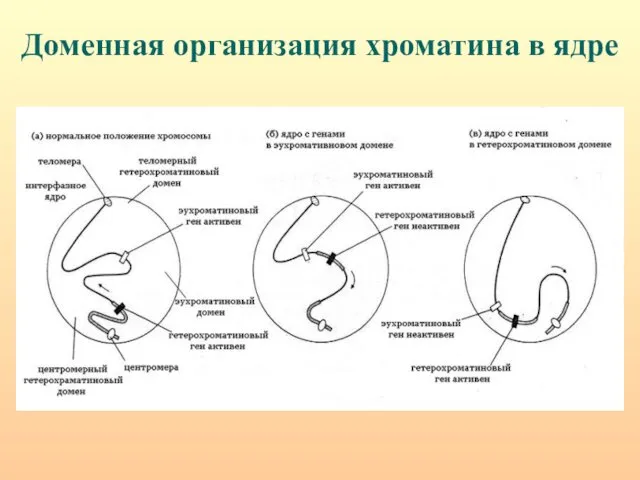
Доменная организация хроматина в ядре
Доменная организация хроматина в ядре
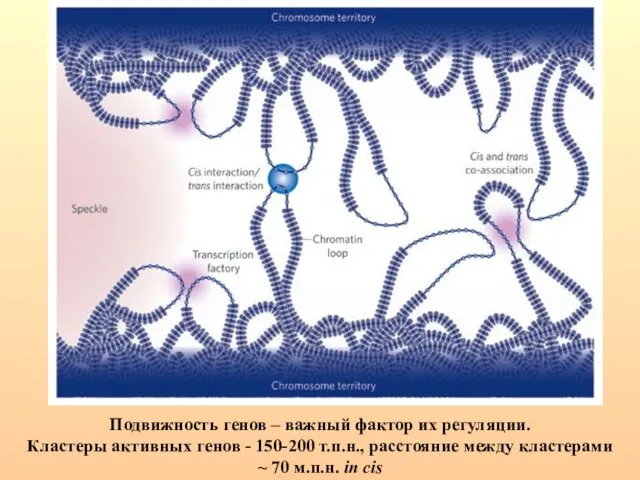
Подвижность генов – важный фактор их регуляции.
Кластеры активных генов - 150-200
Подвижность генов – важный фактор их регуляции.
Кластеры активных генов - 150-200
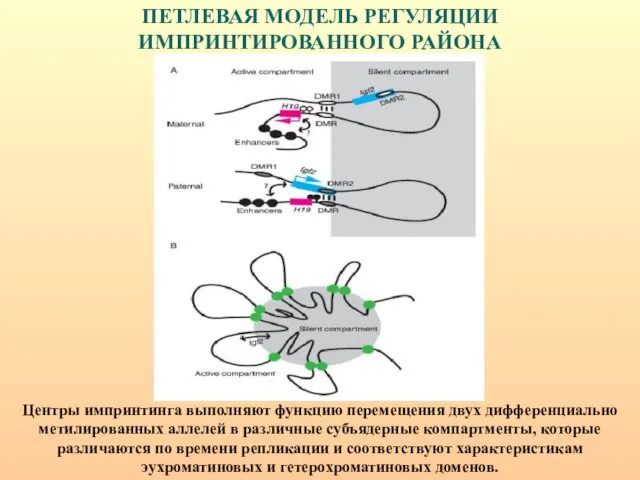
ПЕТЛЕВАЯ МОДЕЛЬ РЕГУЛЯЦИИ ИМПРИНТИРОВАННОГО РАЙОНА
Центры импринтинга выполняют функцию перемещения двух дифференциально
ПЕТЛЕВАЯ МОДЕЛЬ РЕГУЛЯЦИИ ИМПРИНТИРОВАННОГО РАЙОНА
Центры импринтинга выполняют функцию перемещения двух дифференциально
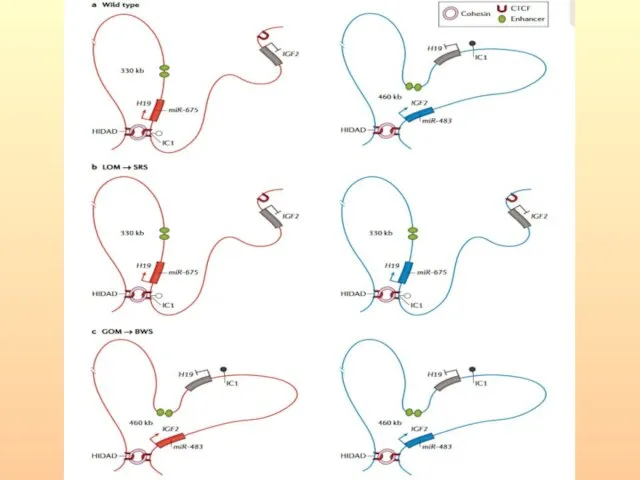




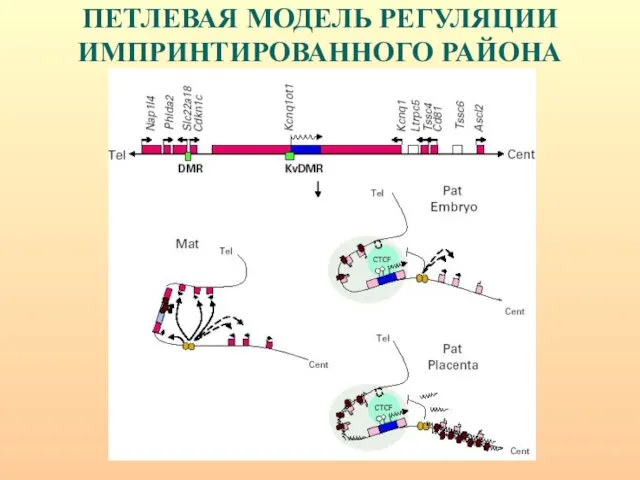
ПЕТЛЕВАЯ МОДЕЛЬ РЕГУЛЯЦИИ ИМПРИНТИРОВАННОГО РАЙОНА
ПЕТЛЕВАЯ МОДЕЛЬ РЕГУЛЯЦИИ ИМПРИНТИРОВАННОГО РАЙОНА
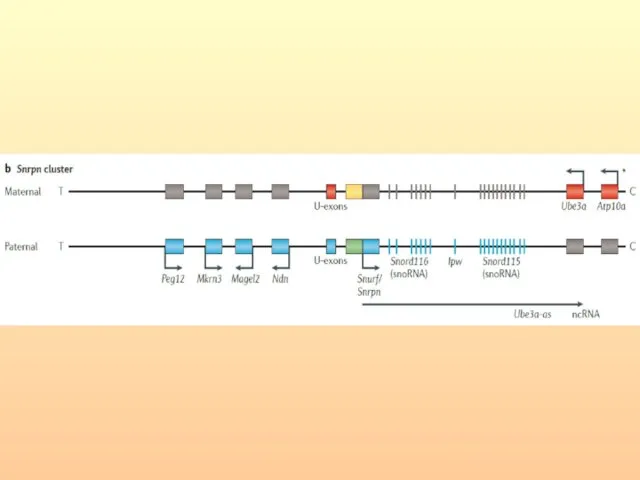
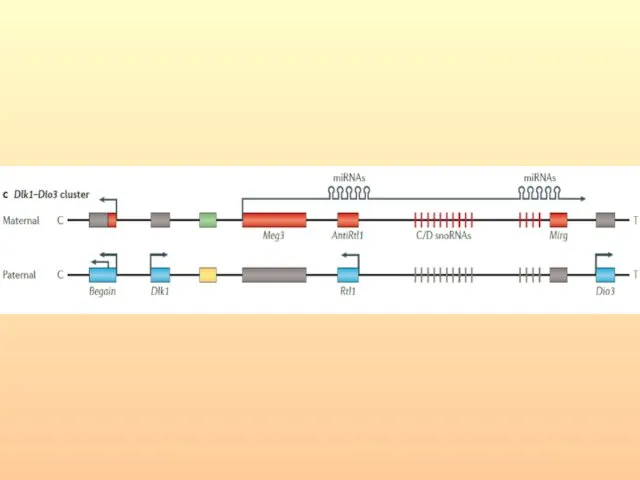
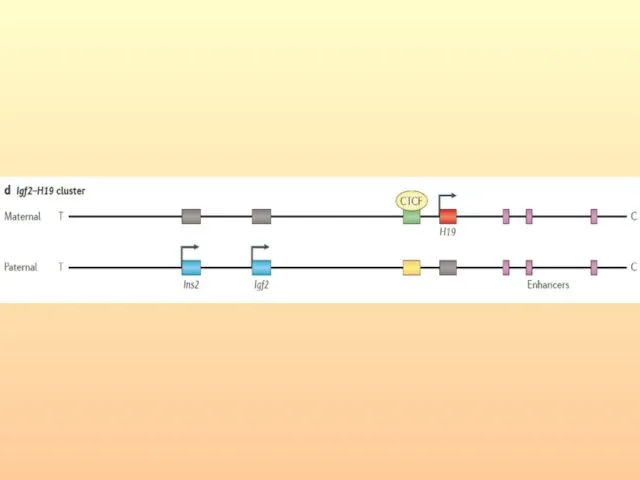
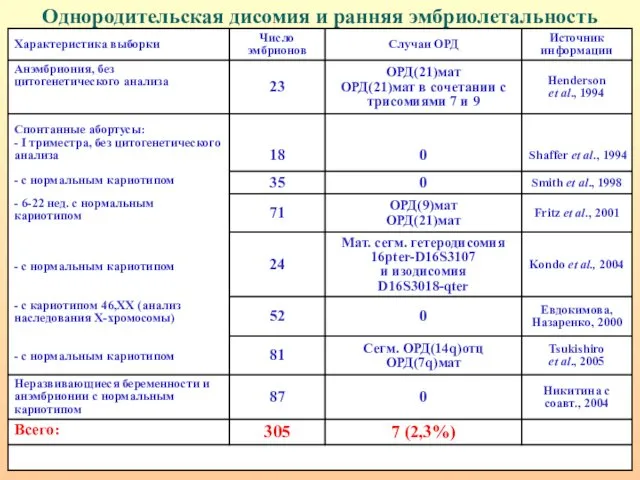
Однородительская дисомия и ранняя эмбриолетальность
Однородительская дисомия и ранняя эмбриолетальность

Целый ряд заболеваний по характеру наследования и проявлениям может возникать вследствие
Целый ряд заболеваний по характеру наследования и проявлениям может возникать вследствие
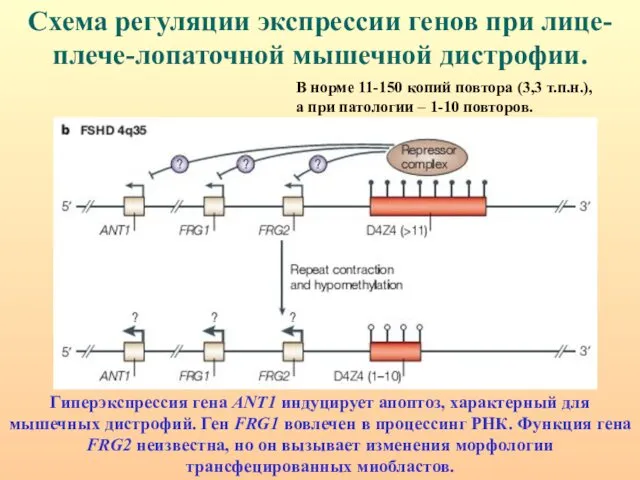
Схема регуляции экспрессии генов при лице-плече-лопаточной мышечной дистрофии.
Гиперэкспрессия гена ANT1
Схема регуляции экспрессии генов при лице-плече-лопаточной мышечной дистрофии.
Гиперэкспрессия гена ANT1
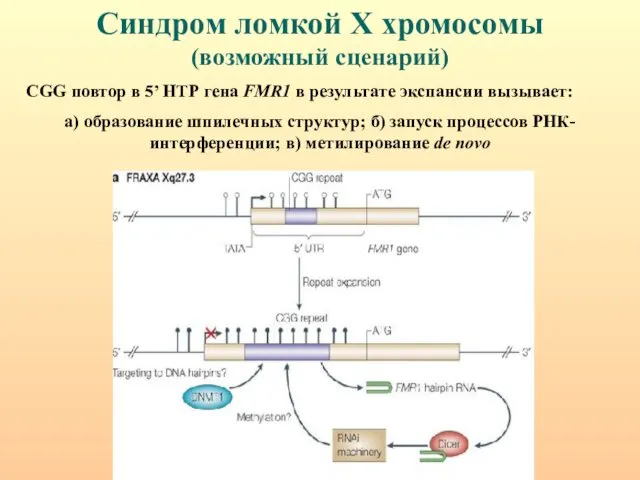
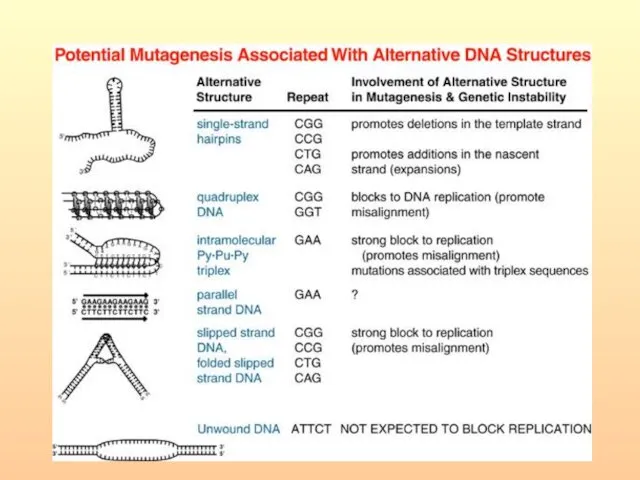
Синдром ломкой Х хромосомы
(возможный сценарий)
CGG повтор в 5’ НТР гена
Синдром ломкой Х хромосомы
(возможный сценарий)
CGG повтор в 5’ НТР гена
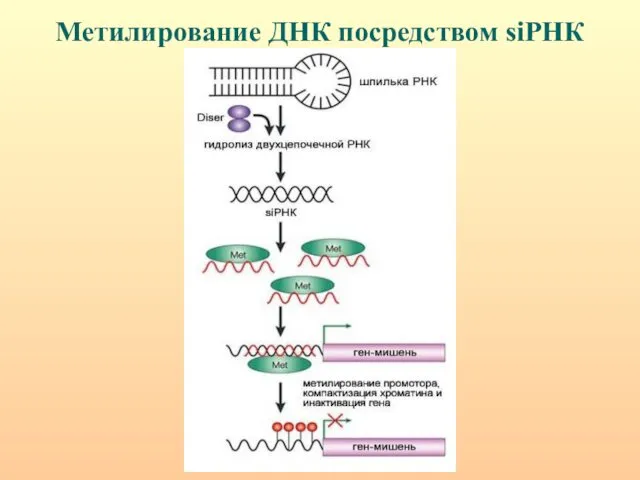
Метилирование ДНК посредством siРНК
Метилирование ДНК посредством siРНК
 Психология пищевого поведения подростков и взрослых
Психология пищевого поведения подростков и взрослых Колоректальный рак
Колоректальный рак Иерархия построения радиотехнической системы
Иерархия построения радиотехнической системы ПРЕЗЕНТАЦИЯ ДЛЯ ВОСПИТАТЕЛЕЙ ТРЕБОВАНИЯ К ОРГАНИЗАЦИИ КНИЖНОГО УГОЛКА
ПРЕЗЕНТАЦИЯ ДЛЯ ВОСПИТАТЕЛЕЙ ТРЕБОВАНИЯ К ОРГАНИЗАЦИИ КНИЖНОГО УГОЛКА Государство Украина
Государство Украина Краеведение - наука о местности, в которой мы живём. Предмет биологического краеведения.
Краеведение - наука о местности, в которой мы живём. Предмет биологического краеведения. Формы организации производства
Формы организации производства С днем рождения, Ксюша
С днем рождения, Ксюша Продолжение Электронное портфолио 23 февраля
Продолжение Электронное портфолио 23 февраля 17.04. Літ. чит. М. Трублаїні Шоколад
17.04. Літ. чит. М. Трублаїні Шоколад Сильвестр Ольшевский, архиепископ Омский
Сильвестр Ольшевский, архиепископ Омский Первичная обр.рыбы
Первичная обр.рыбы Виды и оформление организационно-распорядительной документации
Виды и оформление организационно-распорядительной документации Кубань в каменном веке
Кубань в каменном веке Детёныши животных
Детёныши животных Изображение фигуры человека
Изображение фигуры человека Ф. И. Тютчев. 1803 – 1873 гг. Страницы биографии и творчества
Ф. И. Тютчев. 1803 – 1873 гг. Страницы биографии и творчества Культура речи, как основа эффективного общения
Культура речи, как основа эффективного общения Пищевые связи в экосистеме. Трофические уровни. Типы пищевых цепей
Пищевые связи в экосистеме. Трофические уровни. Типы пищевых цепей Генеральный план. Благоустройство территории
Генеральный план. Благоустройство территории Великобритания на рубеже XIX-XX веков
Великобритания на рубеже XIX-XX веков Легендарный разведчик Николай Иванович Кузнецов
Легендарный разведчик Николай Иванович Кузнецов Кавказские Минеральные Воды
Кавказские Минеральные Воды карточка RGB Диск
карточка RGB Диск Методы медицинской генетики человека
Методы медицинской генетики человека Повышение надежности систем электроснабжения
Повышение надежности систем электроснабжения Древняя Индия
Древняя Индия Явление царя. Подвиг искупления
Явление царя. Подвиг искупления