- Хромосомные болезни человека

Содержание
- 2. СОДЕРЖАНИЕ 1. Генетика человека …………………………………………….3 2. Методы изучения генетики человека …………………….4 3. Наследственные болезни человека ……………………..10
- 3. Генетика человека В 1929 г. советский генетик, невропатолог С.Н.Давиденко организовал первую в мире медико-генетическую консультацию. Он
- 4. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА 1. Клинико-генеалогический метод (составление родословных, предложил в1865 г. Ф.Гальтон). 2. Близнецовый метод
- 5. Клинико-генеалогический метод - здоровый мужчина (больной - ) - здоровая женщина (больная - ) Метод состоит
- 6. Близнецовый метод Двойни встречаются 1/84 новорожденных, 1/3 из них – монозиготные (однояйцовые – близнецы), остальные -
- 7. Коэффициент интеллекта, или IQ, позволяет количественно измерить генетически обусловленные умственные способности людей ( у дизиготных близнецов
- 8. Дерматоглифический метод В генетике используются разделы: дактилоскопия (рис. на подушечках пальцев), пальмоскопия (рис. на ладонях) и
- 9. Цитогенетический метод В 1956 г. швед. ученые Д. Тийо и А Левин разработали метод культивирования человеческих
- 10. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ЧЕЛОВЕКА
- 11. Аутосомно-доминирующий тип наследования 1. Болезнь встечается в каждом поколении родословной. 2. Соотношение больных мальчиков и девочек
- 12. МИКРОСОМИЯ Синдром первой жаберной дуги. Клинические признаки: односторонняя аномалия ушной раковины и гипоплазия нижней челюсти; аномалии
- 13. РОБИНОВА СИНДРОМ Впервые описан в 1969 г. Клинические признаки: необычное строение лица, умеренная карликовость, гипоплазия половых
- 14. ВИЛЛЬЯМСА СИНДРОМ Впервые описан в 1961 г. Клинические признаки: Необычное лицо, низкий рост, короткий нос, полные
- 15. МАРФАНА СИНДРОМ Впервые описан в 1896 г. Клинические признаки: высокий рост, арахнодактилия, подвывих хрусталика, порок митрального
- 16. Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от аневризма аорты. Единственная компенсация –
- 17. Акроцефалосиндактилия Клинические признаки: изменение черепа, гипоплазия основания черепа, плоский лоб, гипертелоризм, запавшая переносица, синдактилия, косоглазие, слабоумие.
- 18. Трихо-рино-фалангетальный синдром Клинические признаки: отставание в росте, лицо с грушевидным носом, оттопыренные уши, редкие, тонкие и
- 19. ПОЛИДАКТИЛИЯ Клинические признаки: существует два варианта: тип А, при котором дополнительный палец функционален, и тип В,
- 20. СИНДАКТИЛИЯ Клинические признаки: синдактилия – это сращение различных пальцев кистей и стоп. На кистях чаще всего
- 21. ОСТЕОГЕНЕЗ Клинические признаки: повышенная ломкость трубчатых костей, ребер и ключиц при минимальной травме, деформации конечностей, голубые
- 22. МИОТОНИЧЕСКАЯ ДИСТРОФИЯ Миотоническая дистрофия, или болезнь Штейнерта – многосистемное заболевание у обоих полов. Клинические признаки: миотония,
- 23. ЭКТРОДАКТИЛИЯ Впервые описан в 1970 г. Клинические признаки: недоразвитие или отсутствие одного или нескольких пальцев кистей
- 24. СИНДРОМ КРУЗОНА (черепно-лицевой дизостоз) Синдром Крузона – дефект гена каспазы, 10q. Впервые описан в 1912 г.
- 25. АХОНДРОПЛАЗИЯ Клинические признаки: диспропорциональная карликовость (рост 120-130 см) за счет укорочения конечностей, большой череп, кисти широкие
- 26. ВИТИЛИГО Клинические признаки: частичная депигментация кожи; поражение обычно симметричное на руках, лице, шее. Больные очень чувствительны
- 27. ГИПЕРТРИХОЗ («ЛЮДИ – ВОЛКИ») Клинические признаки: чрезмерный рост волос на всех частях тела, кроме ладоней и
- 28. Порфирия, или вампиризм "..Ученые выяснили, что вампиризм – это тяжелое, очень редкое заболевание – порфирия, которая
- 29. Аутосомно-рецессивный тип наследования 1. Больной ребенок рождается у клинически здоровых родителей. 2. Болеют сибсы, т.е. братья
- 30. АХОНДРОГЕНЕЗ Клинические признаки: водянка плода, резкое укорочение конечностей, шеи и туловища, большие размеры черепа. Рентгенологически выявляется
- 31. Лоуренса-Муна-Барде-Бидля синдром Впервые описан в 1866 г. J. Laurence и R. Moon. Клинические признаки: жирение, гипогонадизм,
- 32. АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ Клинические признаки: женский псевдогермафродитизм, повышенная секреция гормонов коры надпочечников; гипертрофия клитора и гиперпигментация генитальной
- 33. РАСЩЕЛИНА ГУБЫ Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, часто эпикант и телоризм, деформации первых пальцев
- 34. ЧЕРЕП В ФОРМЕ ТРИЛИСТНИКА Клинические признаки: характерная форма черепа (возникает вследствие внутриутробного зарастания швов) и лица,
- 35. НУНАН СИНДРОМ Впервые описан в 1928 г. Клинические признаки: гипертелоризм, эпикант, низко посаженные уши, нарушение прикуса,
- 36. КОККЕЙНА СИНДРОМ Впервые описан в 1946 г. Клинические признаки: низкорослость, старообразное лицо, микроцефалия, умствен - ная
- 37. КСЕРОДЕРМА ПИГМЕНТНАЯ (дерматоз Капоши) Пигментная ксеродерма – заболевание, протекающее с поражением кожи, фоточувствительностью, злокачественными новообразованиями. Клинические
- 38. ФЕНИЛКЕТОНУРИЯ Фенилкетонурия – болезнь аминокислотного обмена. Описана в 1934 г. А. Фелингом. Патология связана с недостаточностью
- 39. МУКОПОЛИСАХАРИДОЗ Синдром Моркио описан в 1929 г. Клинические признаки: отставание в росте, деформация позвоночника и грудины,
- 40. ХРОМОСОМНЫЕ БОЛЕЗНИ Хромосомные заболевания связаны с аномалиями числа или структуры хромосом. Для них характерно: малый рост
- 41. РОДОСЛОВНАЯ С Х-СЦЕПЛЕННЫМ ТИПОМ НАСЛЕДОВАНИЯ 1. Болеют только мальчики по линии матери. 2. Родители пробанда здоровы.
- 42. ГИДРОЦЕФАЛИЯ Клинические признаки: увеличение объема головы, расширение желудочков мозга; истончение и расхождение костей черепа, диспропорция мозговой
- 43. ГЕМОФИЛИЯ А Клинические признаки: под- и внутри кожные кровотечения, кровоизлияния в крупные суставы, подкожные и межмышечные
- 44. СИНДРОМ ДАУНА (ТРИСОМИЯ 21) Описан в 1866 г. Клинические признаки: умственная отсталость, плоское лицо, монголоид -ный
- 45. СИНДРОМ МАРТИНА-БЕЛЛА Синдром Мартина-Белла – самая распространенная (после болезни Дауна) форма умственной отсталости. Мальчики болеют в
- 46. СИНДРОМ ААРСКОГО Синдром Аарского, или лице-пальце-генитальный синдром подобно описан в 1970 г. Клинические признаки: отставание в
- 47. СИНДРОМ КОФФИНА-ЛОУРИ Синдром впервые описан в 1966 г. Клинические признаки: антимонголоидный разрез глаз, гипертелоризм, луковицеобразный нос,
- 48. Синдром трисомии 9р Клинические признаки: умственная отсталость, задержка роста, микробрахице-фалия, антимонголоидный разрез глаз, глубоко посаженные глаза
- 49. СИНДРОМ КЛАЙНФЕЛЬТЕРА (47, ХХУ) Описан в 1942 г. Клинические признаки: высокий рост, хрупкое телосложение, гипоплазия яичек,
- 50. СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА (ХО –СИНДРОМ) Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые вторичные половые признаки, крыловидные
- 51. СИНДРОМ ПАТАУ (ТРИСОМИЯ 13) Описан в 1961 г. Клинические признаки: микроцефалия, расщепле- ние губы и неба,
- 52. Синдром Эдвардса – трисомия 18 Клинические признаки: задержка пренатального развития, множественные пороки развития черепа (маленькая нижняя
- 53. СИНДРОМ КОШАЧЬЕГО КРИКА (МОНОСОМИЯ 5р) Описан в 1963 г. Клинические признаки: необычный плач, напоминающий кошачье мяуканье,
- 54. СИНДРОМ СВАЕРА (ДИСГЕНЕЗИЯ ГОНАД, ХУ ТИП ) Клинические признаки: наружные половые органы сформированы по женскому типу,
- 55. ГЕРМАФРОДИТИЗМ Гермафродитизм у человека— врожденный порок развития, характеризующийся наличием мужских и женских половых признаков одновременно. При
- 56. СИНДРОМ ЭЛЕРСА-ДАНЛО Описан в 1657 г. Клинические признаки: гиперрастяжимость соединительной ткани (нарушение синтеза коллагена); кожа тонкая
- 57. ПРОГЕРИЯ Описана в 1886 г. Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения в 8-10 раз.
- 58. ТЕРМИНОЛОГИЧЕСКИЙ СЛОВАРЬ Акроцефалия - высокий «башенный» череп. Алопеция – стойкое или временное выпадение волос. Аменорея –
- 59. ТЕРМИНОЛОГИЧЕСКИЙ СЛОВАРЬ Прогерия – преждевремен -ное старение организма. Птеригиум – крыловидные складки кожи. Птоз – опущение
- 61. Скачать презентацию

СОДЕРЖАНИЕ
1. Генетика человека …………………………………………….3
2. Методы изучения генетики человека …………………….4
3. Наследственные болезни
СОДЕРЖАНИЕ
1. Генетика человека …………………………………………….3
2. Методы изучения генетики человека …………………….4
3. Наследственные болезни

Генетика человека
В 1929 г. советский генетик, невропатолог С.Н.Давиденко организовал первую в
Генетика человека
В 1929 г. советский генетик, невропатолог С.Н.Давиденко организовал первую в

МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
1. Клинико-генеалогический метод (составление родословных, предложил в1865 г.
МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
1. Клинико-генеалогический метод (составление родословных, предложил в1865 г.
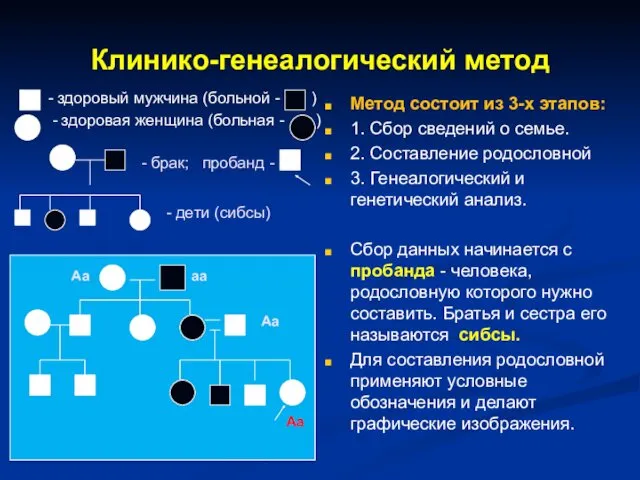
Клинико-генеалогический метод
- здоровый мужчина (больной - )
- здоровая женщина
Клинико-генеалогический метод
- здоровый мужчина (больной - )
- здоровая женщина

Близнецовый метод
Двойни встречаются 1/84 новорожденных, 1/3 из них – монозиготные (однояйцовые
Близнецовый метод
Двойни встречаются 1/84 новорожденных, 1/3 из них – монозиготные (однояйцовые

Коэффициент интеллекта, или IQ, позволяет количественно измерить генетически обусловленные умственные способности
Коэффициент интеллекта, или IQ, позволяет количественно измерить генетически обусловленные умственные способности
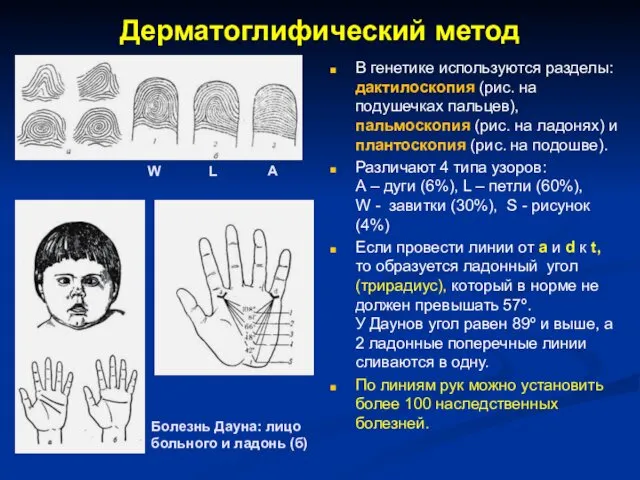
Дерматоглифический метод
В генетике используются разделы: дактилоскопия (рис. на подушечках пальцев), пальмоскопия
Дерматоглифический метод
В генетике используются разделы: дактилоскопия (рис. на подушечках пальцев), пальмоскопия

Цитогенетический метод
В 1956 г. швед. ученые Д. Тийо и А Левин
Цитогенетический метод
В 1956 г. швед. ученые Д. Тийо и А Левин
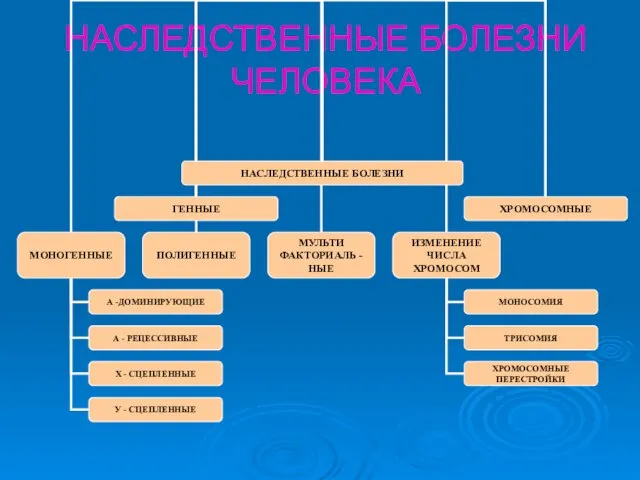
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ЧЕЛОВЕКА
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ЧЕЛОВЕКА
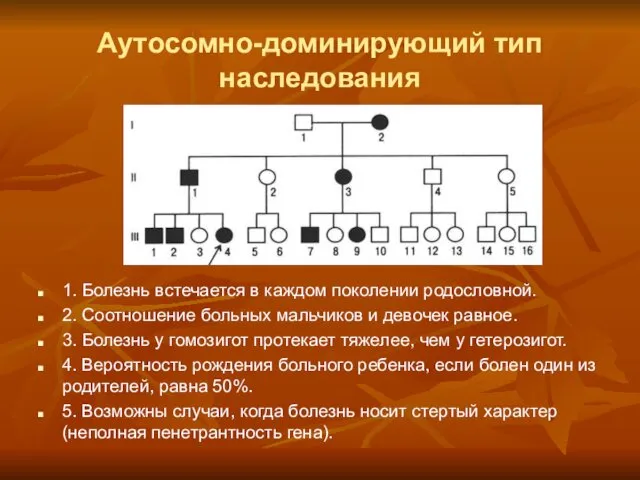
Аутосомно-доминирующий тип наследования
1. Болезнь встечается в каждом поколении родословной.
2. Соотношение больных
Аутосомно-доминирующий тип наследования
1. Болезнь встечается в каждом поколении родословной.
2. Соотношение больных
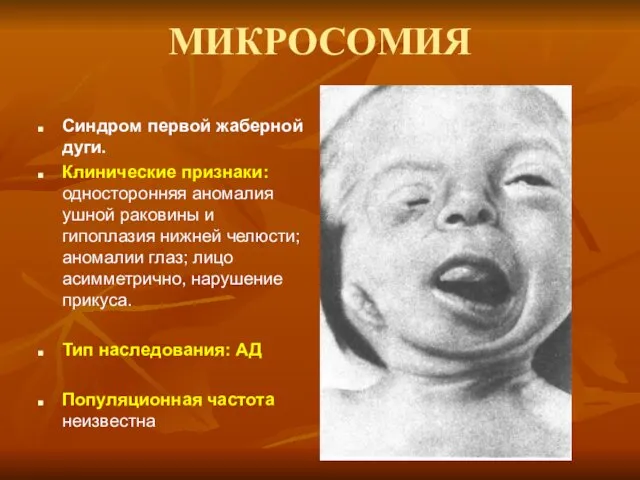
МИКРОСОМИЯ
Синдром первой жаберной дуги.
Клинические признаки: односторонняя аномалия ушной раковины и гипоплазия
МИКРОСОМИЯ
Синдром первой жаберной дуги.
Клинические признаки: односторонняя аномалия ушной раковины и гипоплазия
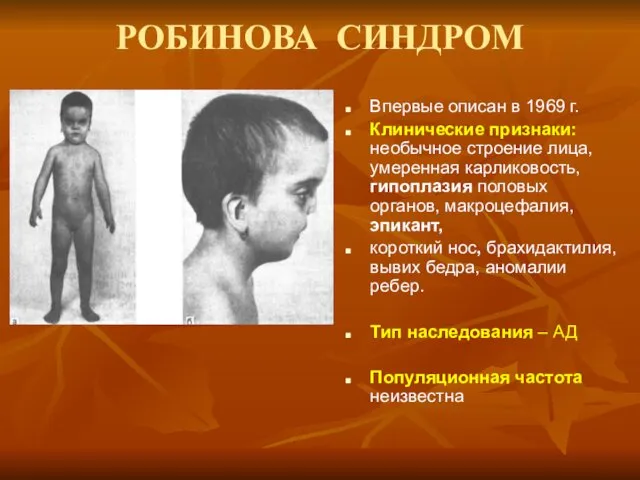
РОБИНОВА СИНДРОМ
Впервые описан в 1969 г.
Клинические признаки: необычное строение лица,
РОБИНОВА СИНДРОМ
Впервые описан в 1969 г.
Клинические признаки: необычное строение лица,

ВИЛЛЬЯМСА СИНДРОМ
Впервые описан в 1961 г.
Клинические признаки:
Необычное лицо, низкий рост, короткий
ВИЛЛЬЯМСА СИНДРОМ
Впервые описан в 1961 г.
Клинические признаки:
Необычное лицо, низкий рост, короткий
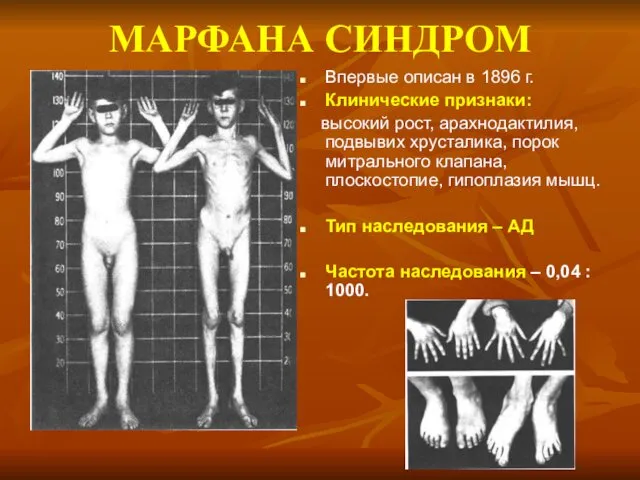
МАРФАНА СИНДРОМ
Впервые описан в 1896 г.
Клинические признаки:
высокий рост, арахнодактилия, подвывих
МАРФАНА СИНДРОМ
Впервые описан в 1896 г.
Клинические признаки:
высокий рост, арахнодактилия, подвывих

Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от
Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от
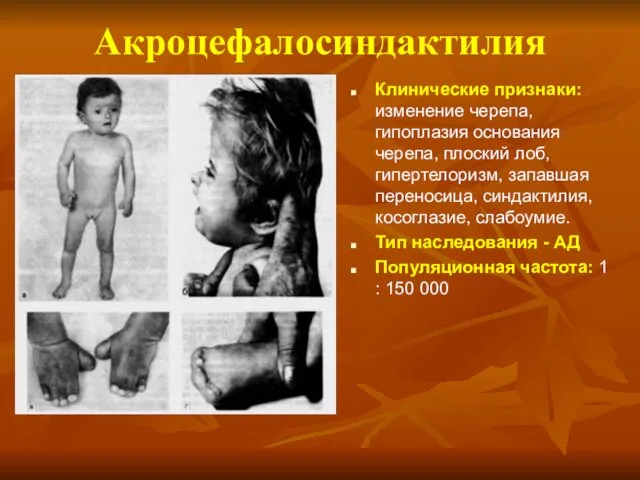
Акроцефалосиндактилия
Клинические признаки: изменение черепа, гипоплазия основания черепа, плоский лоб, гипертелоризм, запавшая
Акроцефалосиндактилия
Клинические признаки: изменение черепа, гипоплазия основания черепа, плоский лоб, гипертелоризм, запавшая

Трихо-рино-фалангетальный синдром
Клинические признаки: отставание в росте, лицо с грушевидным носом, оттопыренные
Трихо-рино-фалангетальный синдром
Клинические признаки: отставание в росте, лицо с грушевидным носом, оттопыренные
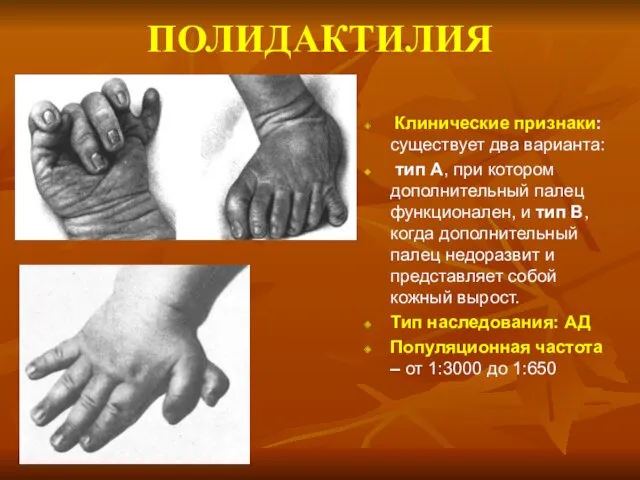
ПОЛИДАКТИЛИЯ
Клинические признаки: существует два варианта:
тип А, при котором дополнительный
ПОЛИДАКТИЛИЯ
Клинические признаки: существует два варианта:
тип А, при котором дополнительный
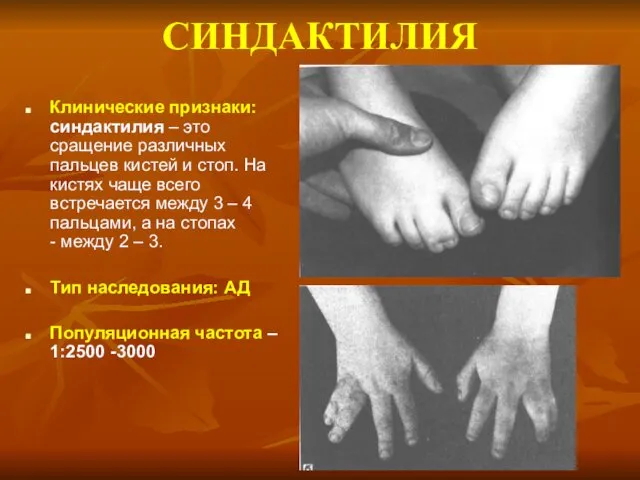
СИНДАКТИЛИЯ
Клинические признаки: синдактилия – это сращение различных пальцев кистей и стоп.
СИНДАКТИЛИЯ
Клинические признаки: синдактилия – это сращение различных пальцев кистей и стоп.
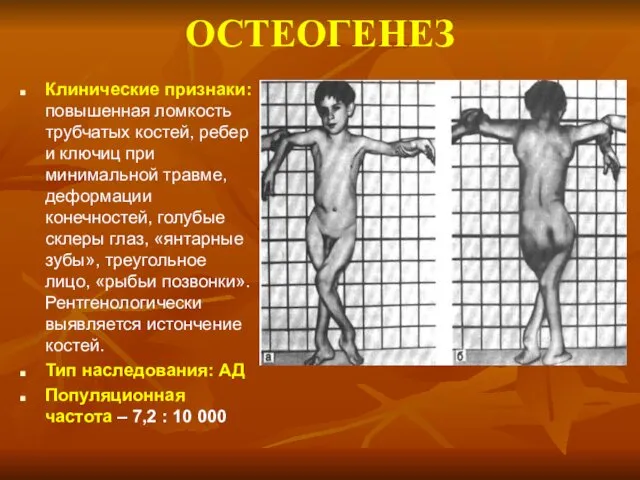
ОСТЕОГЕНЕЗ
Клинические признаки: повышенная ломкость трубчатых костей, ребер и ключиц при минимальной
ОСТЕОГЕНЕЗ
Клинические признаки: повышенная ломкость трубчатых костей, ребер и ключиц при минимальной

МИОТОНИЧЕСКАЯ ДИСТРОФИЯ
Миотоническая дистрофия, или болезнь Штейнерта – многосистемное заболевание у обоих
МИОТОНИЧЕСКАЯ ДИСТРОФИЯ
Миотоническая дистрофия, или болезнь Штейнерта – многосистемное заболевание у обоих

ЭКТРОДАКТИЛИЯ
Впервые описан в 1970 г.
Клинические признаки: недоразвитие или отсутствие одного или
ЭКТРОДАКТИЛИЯ
Впервые описан в 1970 г.
Клинические признаки: недоразвитие или отсутствие одного или
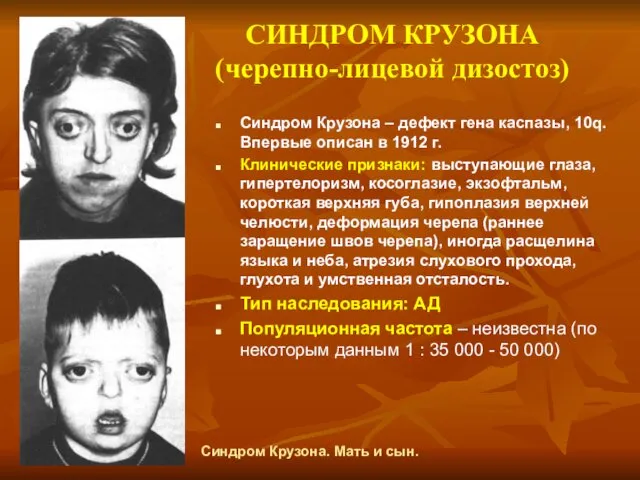
СИНДРОМ КРУЗОНА (черепно-лицевой дизостоз)
Синдром Крузона – дефект гена каспазы, 10q. Впервые
СИНДРОМ КРУЗОНА (черепно-лицевой дизостоз)
Синдром Крузона – дефект гена каспазы, 10q. Впервые
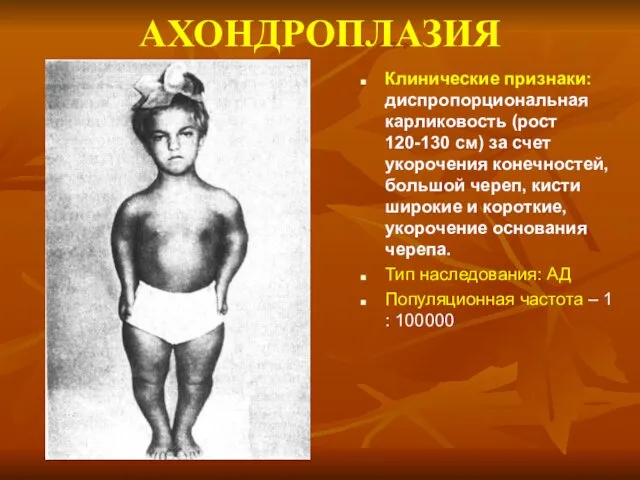
АХОНДРОПЛАЗИЯ
Клинические признаки: диспропорциональная карликовость (рост 120-130 см) за счет укорочения конечностей,
АХОНДРОПЛАЗИЯ
Клинические признаки: диспропорциональная карликовость (рост 120-130 см) за счет укорочения конечностей,
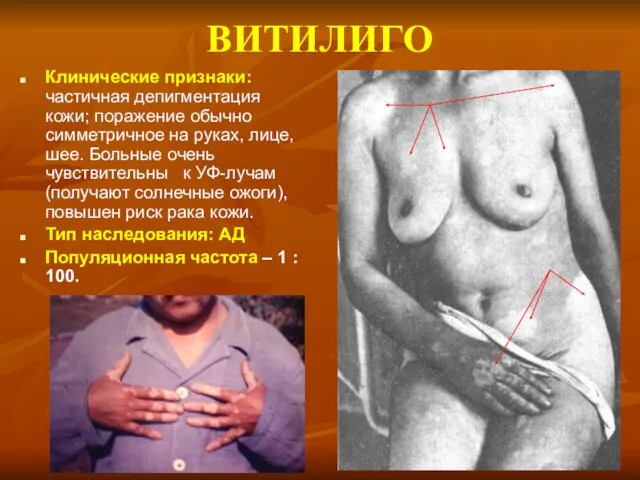
ВИТИЛИГО
Клинические признаки: частичная депигментация кожи; поражение обычно симметричное на руках, лице,
ВИТИЛИГО
Клинические признаки: частичная депигментация кожи; поражение обычно симметричное на руках, лице,

ГИПЕРТРИХОЗ («ЛЮДИ – ВОЛКИ»)
Клинические признаки: чрезмерный рост волос на всех частях
ГИПЕРТРИХОЗ («ЛЮДИ – ВОЛКИ»)
Клинические признаки: чрезмерный рост волос на всех частях

Порфирия, или вампиризм
"..Ученые выяснили, что вампиризм – это тяжелое, очень редкое
Порфирия, или вампиризм
"..Ученые выяснили, что вампиризм – это тяжелое, очень редкое
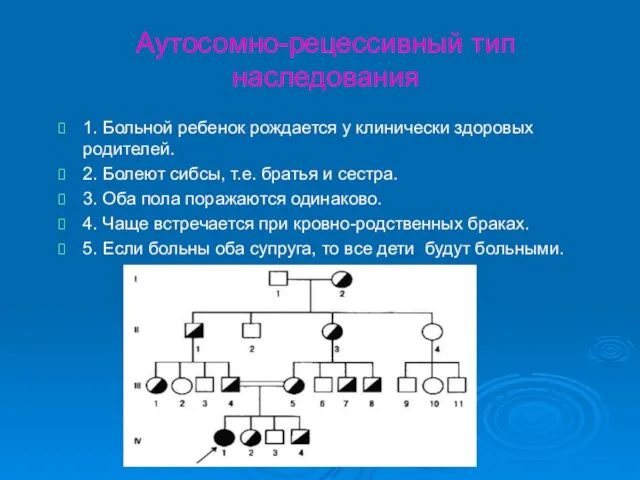
Аутосомно-рецессивный тип наследования
1. Больной ребенок рождается у клинически здоровых родителей.
2. Болеют
Аутосомно-рецессивный тип наследования
1. Больной ребенок рождается у клинически здоровых родителей.
2. Болеют
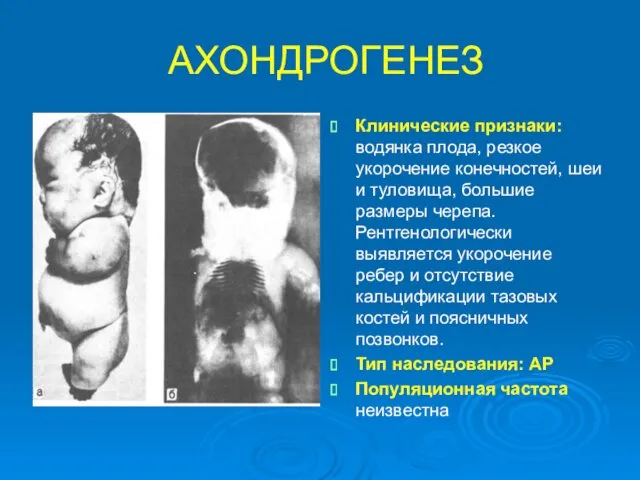
АХОНДРОГЕНЕЗ
Клинические признаки: водянка плода, резкое укорочение конечностей, шеи и туловища, большие
АХОНДРОГЕНЕЗ
Клинические признаки: водянка плода, резкое укорочение конечностей, шеи и туловища, большие
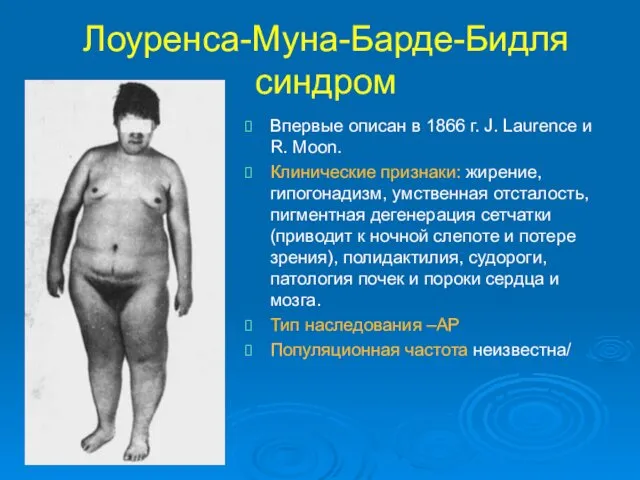
Лоуренса-Муна-Барде-Бидля синдром
Впервые описан в 1866 г. J. Laurence и R. Moon.
Клинические
Лоуренса-Муна-Барде-Бидля синдром
Впервые описан в 1866 г. J. Laurence и R. Moon.
Клинические
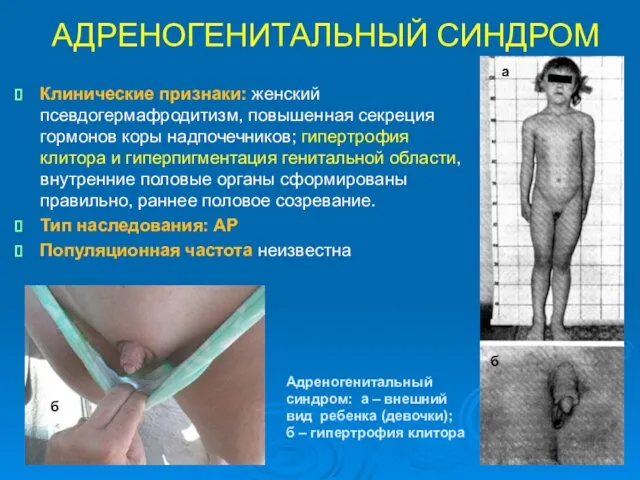
АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ
Клинические признаки: женский псевдогермафродитизм, повышенная секреция гормонов коры надпочечников; гипертрофия
АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ
Клинические признаки: женский псевдогермафродитизм, повышенная секреция гормонов коры надпочечников; гипертрофия
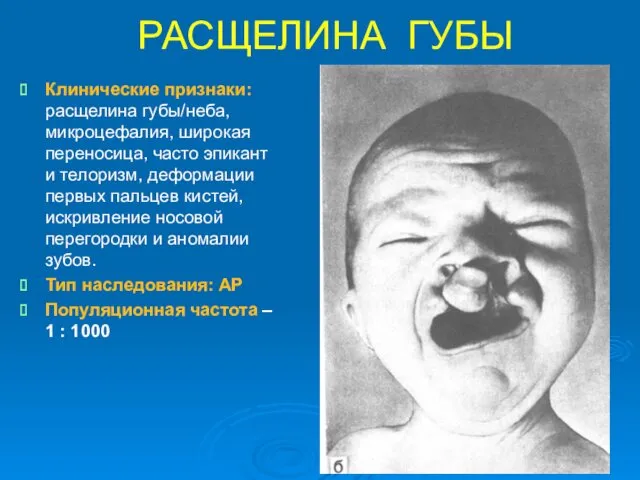
РАСЩЕЛИНА ГУБЫ
Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, часто эпикант и
РАСЩЕЛИНА ГУБЫ
Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, часто эпикант и

ЧЕРЕП В ФОРМЕ ТРИЛИСТНИКА
Клинические признаки: характерная форма черепа (возникает вследствие внутриутробного
ЧЕРЕП В ФОРМЕ ТРИЛИСТНИКА
Клинические признаки: характерная форма черепа (возникает вследствие внутриутробного

НУНАН СИНДРОМ
Впервые описан в 1928 г.
Клинические признаки: гипертелоризм, эпикант, низко
НУНАН СИНДРОМ
Впервые описан в 1928 г.
Клинические признаки: гипертелоризм, эпикант, низко

КОККЕЙНА СИНДРОМ
Впервые описан в 1946 г.
Клинические признаки: низкорослость, старообразное лицо, микроцефалия,
КОККЕЙНА СИНДРОМ
Впервые описан в 1946 г.
Клинические признаки: низкорослость, старообразное лицо, микроцефалия,
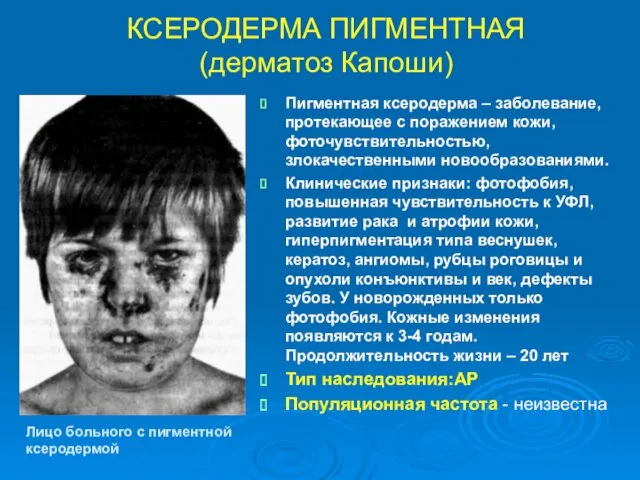
КСЕРОДЕРМА ПИГМЕНТНАЯ (дерматоз Капоши)
Пигментная ксеродерма – заболевание, протекающее с поражением кожи,
КСЕРОДЕРМА ПИГМЕНТНАЯ (дерматоз Капоши)
Пигментная ксеродерма – заболевание, протекающее с поражением кожи,

ФЕНИЛКЕТОНУРИЯ
Фенилкетонурия – болезнь аминокислотного обмена. Описана в 1934 г. А. Фелингом.
ФЕНИЛКЕТОНУРИЯ
Фенилкетонурия – болезнь аминокислотного обмена. Описана в 1934 г. А. Фелингом.
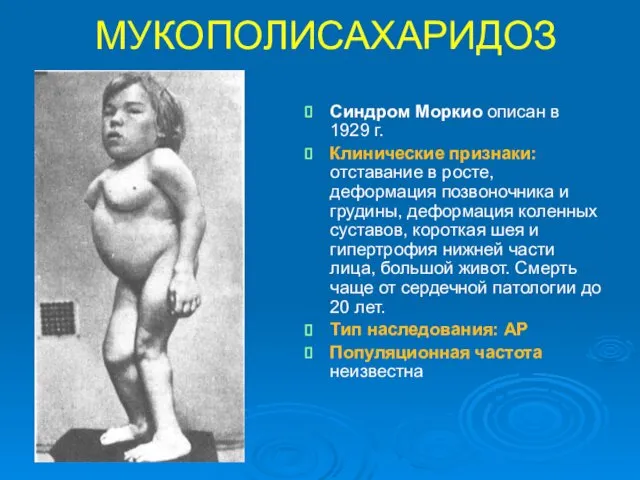
МУКОПОЛИСАХАРИДОЗ
Синдром Моркио описан в 1929 г.
Клинические признаки: отставание в росте, деформация
МУКОПОЛИСАХАРИДОЗ
Синдром Моркио описан в 1929 г.
Клинические признаки: отставание в росте, деформация

ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомные заболевания связаны с аномалиями числа или структуры хромосом.
Для них
ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомные заболевания связаны с аномалиями числа или структуры хромосом.
Для них
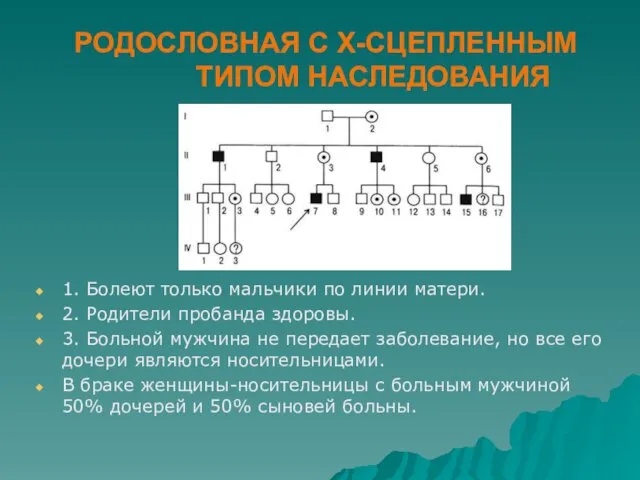
РОДОСЛОВНАЯ С Х-СЦЕПЛЕННЫМ ТИПОМ НАСЛЕДОВАНИЯ
1. Болеют только мальчики по линии
РОДОСЛОВНАЯ С Х-СЦЕПЛЕННЫМ ТИПОМ НАСЛЕДОВАНИЯ
1. Болеют только мальчики по линии

ГИДРОЦЕФАЛИЯ
Клинические признаки: увеличение объема головы, расширение желудочков мозга; истончение и расхождение
ГИДРОЦЕФАЛИЯ
Клинические признаки: увеличение объема головы, расширение желудочков мозга; истончение и расхождение
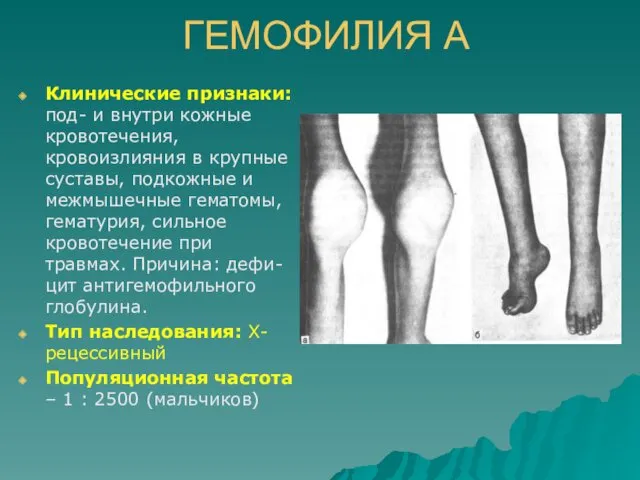
ГЕМОФИЛИЯ А
Клинические признаки: под- и внутри кожные кровотечения, кровоизлияния в крупные
ГЕМОФИЛИЯ А
Клинические признаки: под- и внутри кожные кровотечения, кровоизлияния в крупные
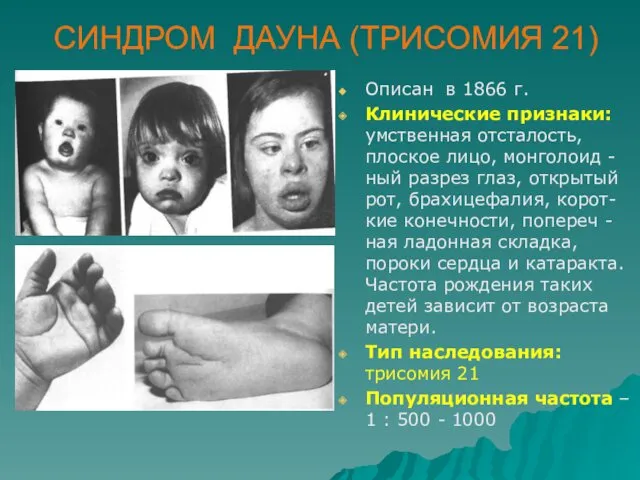
СИНДРОМ ДАУНА (ТРИСОМИЯ 21)
Описан в 1866 г.
Клинические признаки: умственная отсталость, плоское
СИНДРОМ ДАУНА (ТРИСОМИЯ 21)
Описан в 1866 г.
Клинические признаки: умственная отсталость, плоское
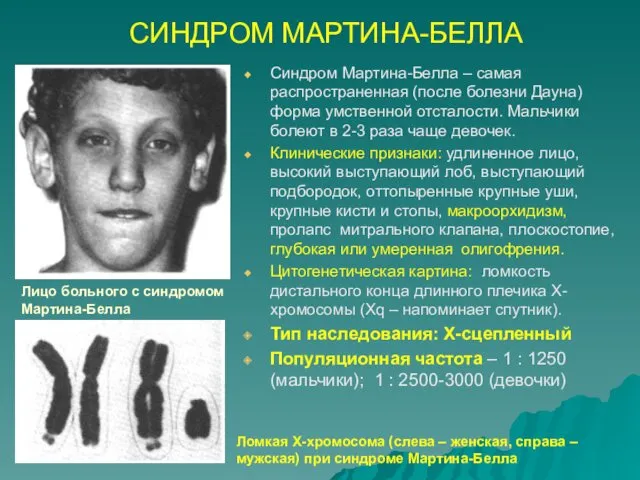
СИНДРОМ МАРТИНА-БЕЛЛА
Синдром Мартина-Белла – самая распространенная (после болезни Дауна) форма умственной
СИНДРОМ МАРТИНА-БЕЛЛА
Синдром Мартина-Белла – самая распространенная (после болезни Дауна) форма умственной
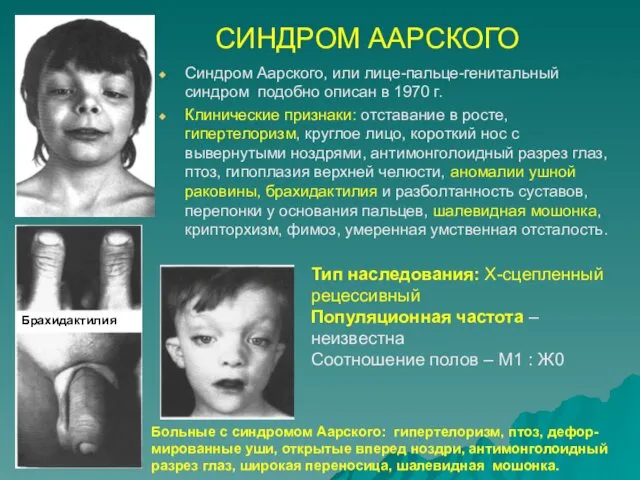
СИНДРОМ ААРСКОГО
Синдром Аарского, или лице-пальце-генитальный синдром подобно описан в 1970 г.
Клинические
СИНДРОМ ААРСКОГО
Синдром Аарского, или лице-пальце-генитальный синдром подобно описан в 1970 г.
Клинические

СИНДРОМ КОФФИНА-ЛОУРИ
Синдром впервые описан в 1966 г.
Клинические признаки: антимонголоидный разрез глаз,
СИНДРОМ КОФФИНА-ЛОУРИ
Синдром впервые описан в 1966 г.
Клинические признаки: антимонголоидный разрез глаз,
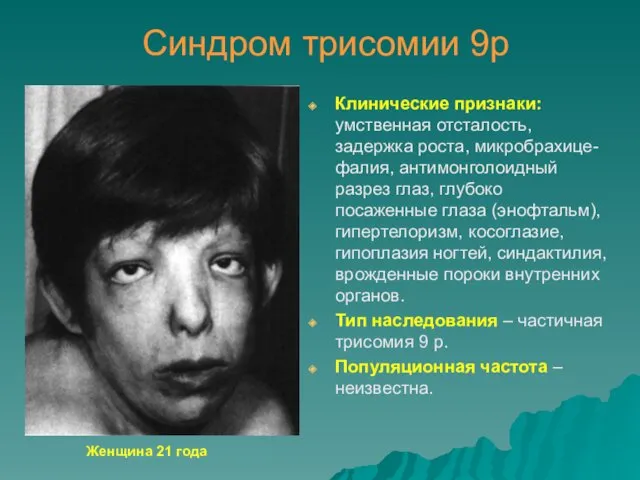
Синдром трисомии 9р
Клинические признаки: умственная отсталость, задержка роста, микробрахице-фалия, антимонголоидный разрез
Синдром трисомии 9р
Клинические признаки: умственная отсталость, задержка роста, микробрахице-фалия, антимонголоидный разрез

СИНДРОМ КЛАЙНФЕЛЬТЕРА (47, ХХУ)
Описан в 1942 г.
Клинические признаки: высокий рост, хрупкое
СИНДРОМ КЛАЙНФЕЛЬТЕРА (47, ХХУ)
Описан в 1942 г.
Клинические признаки: высокий рост, хрупкое

СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА (ХО –СИНДРОМ)
Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые
СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА (ХО –СИНДРОМ)
Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые
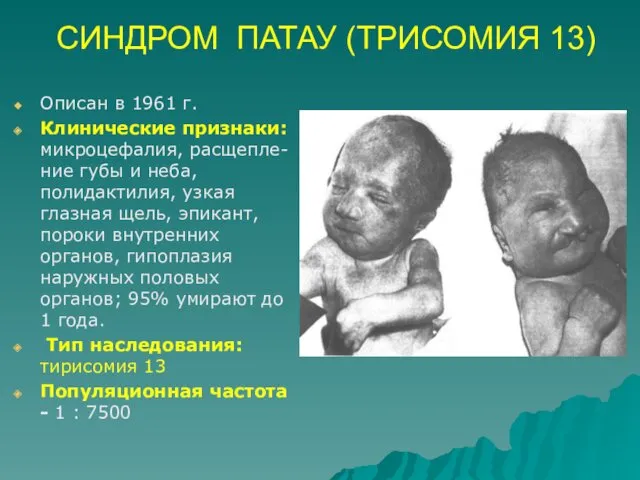
СИНДРОМ ПАТАУ (ТРИСОМИЯ 13)
Описан в 1961 г.
Клинические признаки: микроцефалия, расщепле- ние
СИНДРОМ ПАТАУ (ТРИСОМИЯ 13)
Описан в 1961 г.
Клинические признаки: микроцефалия, расщепле- ние
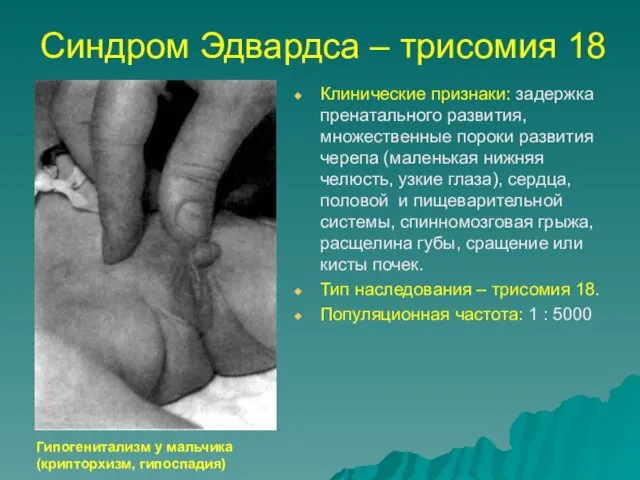
Синдром Эдвардса – трисомия 18
Клинические признаки: задержка пренатального развития, множественные пороки
Синдром Эдвардса – трисомия 18
Клинические признаки: задержка пренатального развития, множественные пороки
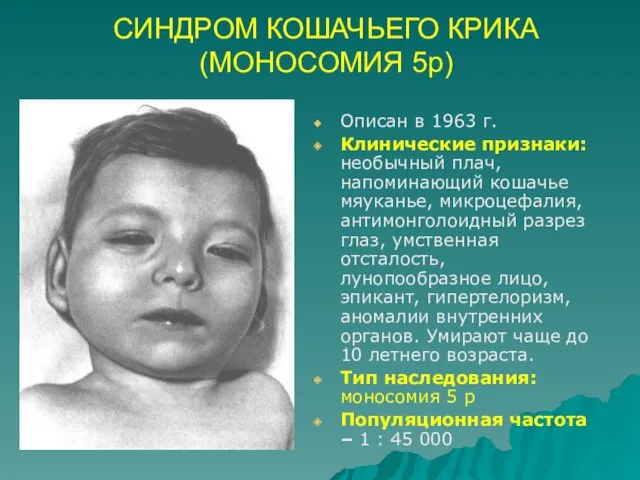
СИНДРОМ КОШАЧЬЕГО КРИКА (МОНОСОМИЯ 5р)
Описан в 1963 г.
Клинические признаки: необычный плач,
СИНДРОМ КОШАЧЬЕГО КРИКА (МОНОСОМИЯ 5р)
Описан в 1963 г.
Клинические признаки: необычный плач,

СИНДРОМ СВАЕРА (ДИСГЕНЕЗИЯ ГОНАД, ХУ ТИП )
Клинические признаки: наружные половые органы
СИНДРОМ СВАЕРА (ДИСГЕНЕЗИЯ ГОНАД, ХУ ТИП )
Клинические признаки: наружные половые органы

ГЕРМАФРОДИТИЗМ
Гермафродитизм у человека— врожденный порок развития, характеризующийся наличием мужских и женских
ГЕРМАФРОДИТИЗМ
Гермафродитизм у человека— врожденный порок развития, характеризующийся наличием мужских и женских

СИНДРОМ ЭЛЕРСА-ДАНЛО
Описан в 1657 г.
Клинические признаки: гиперрастяжимость соединительной ткани (нарушение синтеза
СИНДРОМ ЭЛЕРСА-ДАНЛО
Описан в 1657 г.
Клинические признаки: гиперрастяжимость соединительной ткани (нарушение синтеза
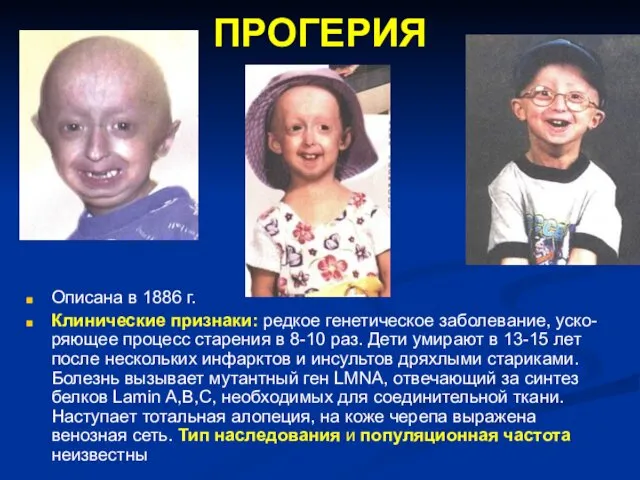
ПРОГЕРИЯ
Описана в 1886 г.
Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения
ПРОГЕРИЯ
Описана в 1886 г.
Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения

ТЕРМИНОЛОГИЧЕСКИЙ СЛОВАРЬ
Акроцефалия - высокий «башенный» череп.
Алопеция – стойкое или временное
ТЕРМИНОЛОГИЧЕСКИЙ СЛОВАРЬ
Акроцефалия - высокий «башенный» череп.
Алопеция – стойкое или временное

ТЕРМИНОЛОГИЧЕСКИЙ СЛОВАРЬ
Прогерия – преждевремен -ное старение организма.
Птеригиум – крыловидные складки кожи.
Птоз
ТЕРМИНОЛОГИЧЕСКИЙ СЛОВАРЬ
Прогерия – преждевремен -ное старение организма.
Птеригиум – крыловидные складки кожи.
Птоз
 Работа с тревожными детьми
Работа с тревожными детьми Индивидуальная теория личности Альфреда Адлера
Индивидуальная теория личности Альфреда Адлера Экономика и особенности деятельности отраслей культуры
Экономика и особенности деятельности отраслей культуры Этапы развития исторического знания. Становление и развитие взглядов на мир. Периодизация всемирной истории
Этапы развития исторического знания. Становление и развитие взглядов на мир. Периодизация всемирной истории Состав числа из двух меньших чисел
Состав числа из двух меньших чисел Взаимосвязь биологических и социальных факторов в психическом развитии
Взаимосвязь биологических и социальных факторов в психическом развитии Pro-файл. Екатерина Смольянова
Pro-файл. Екатерина Смольянова мы против наркотиков
мы против наркотиков Общественное здоровье населения как экономическая категория. Здоровье населения. Медико-социальные аспекты здоровья
Общественное здоровье населения как экономическая категория. Здоровье населения. Медико-социальные аспекты здоровья Презентация проекта Прогулки с интересом
Презентация проекта Прогулки с интересом Последствия курения (кл.час)
Последствия курения (кл.час) История_Духовенство
История_Духовенство Й-Ї
Й-Ї Как путешествует письмо?
Как путешествует письмо? HAICTC el planeta es para nuestros hijos. BIOMASA primera fuente de energías renovables
HAICTC el planeta es para nuestros hijos. BIOMASA primera fuente de energías renovables Номинальные напряжения в системах электроснабжения городов
Номинальные напряжения в системах электроснабжения городов Нормативно-правовые документы и рекомендации, регламентирующие деятельность образовательных организаций
Нормативно-правовые документы и рекомендации, регламентирующие деятельность образовательных организаций Презентация Парциальные программы по театрализованной деятельности детей дошкольного возраста
Презентация Парциальные программы по театрализованной деятельности детей дошкольного возраста Основы оперативной техники
Основы оперативной техники Препарат Райкат старт
Препарат Райкат старт Объектно-ориентированное программирование. Обзор среды разработки Visual Studio .NET
Объектно-ориентированное программирование. Обзор среды разработки Visual Studio .NET Духовно – нравственное и патриотическое воспитание детей дошкольного возраста посредством использования устного народного творчества: фольклор.
Духовно – нравственное и патриотическое воспитание детей дошкольного возраста посредством использования устного народного творчества: фольклор. Юность. Николай Алексеевич Некрасов
Юность. Николай Алексеевич Некрасов Атомные электростанции (АЭС)
Атомные электростанции (АЭС) Единый орфографический режим в начальной школе
Единый орфографический режим в начальной школе Виды биопсий
Виды биопсий Счастливейшая женщина планеты. С юбилеем
Счастливейшая женщина планеты. С юбилеем E-waste
E-waste