- Лимфопролиферативные заболевания

Содержание
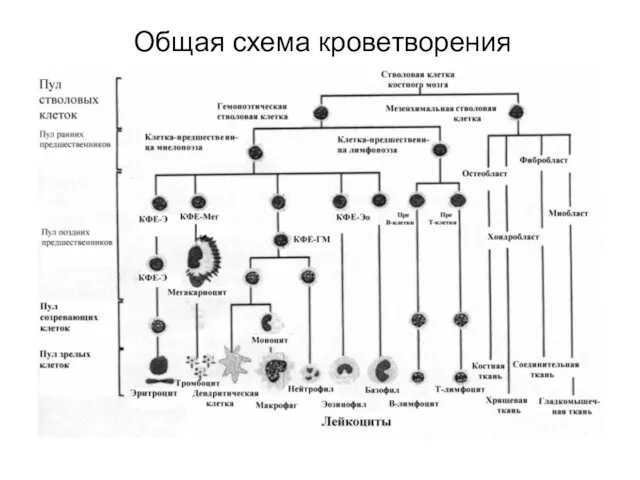
- 2. Общая схема кроветворения
- 3. Этапы созревания лимфоцитов костный мозг стволовая клетка – клетка предшественница В-лимфоцит Т-лимфоцит лимфоузлы тимус селезенка скопления
- 4. Этиопатогенез лимфопролиферативных заболеваний Предрасполагающие факторы: хроническая антигенная стимуляция, иммунодефицит и вирусные инфекции. Первичное онкогенное событие, запускающее
- 5. Генетические повреждения при лимфомах можно подразделить на две крупные категории: активация протоонкогенов и инактивация генов–супрессоров опухолевого
- 6. Хромосомные нарушения при различных НХЛ Транс-локации Прото-онкогены Функции прото- онкогенов лимфома t(14;18) BCL2 Супрессия апоптоза Фолликулярная
- 7. При созревании от костно-мозговой клетки-предшественницы лимфопоэза до плазматической клетки, геном В-лимфоцитов подвергается многочисленным перестройкам в ходе
- 8. перестройки генов Ig, соматической гипермутации, сдвига изотипа из-за ошибок этих механизмов. Изменения генома при лимфомах могут
- 9. Главная особенность лимфомагенеза в преобладании среди хромосомных нарушений транслокаций с участием локусов генов иммуноглобулинов.
- 10. В-клеточные опухоли с фенотипом зрелых лимфоцитов Хронический лимфоцитарный лейкоз / лимфоцитарная лимфома В-клеточный пролимфоцитарный лейкоз Лимфоплазмоцитарная
- 11. Классификация ВОЗ лимфопролиферативных заболеваний (2) В-клеточные лимфопролиферативные процессы с неопределенным опухолевым потенциалом Лимфоматоидный гранулематоз Посттрансплантационное лимфопролиферативное
- 12. В-клеточные лимфопролиферативные заболевания, не вошедшие в классификацию ВОЗ Внутрисосудистая В-крупноклеточная лимфома Первичный амилоидоз (AL) Болезнь тяжёлых
- 13. T- и NK-клеточные опухоли с фенотипом зрелых лимфоцитов Лейкозы и первично диссеминированные лимфомы Т-клеточный пролимфоцитарный лейкоз
- 14. В-клеточный хронический лимфолейкоз - моноклональная пролиферация CD5-позитивных зрелых В-лимфоцитов с первичным поражением костного мозга и вовлечением
- 15. Хронический лимфолейкоз Наиболее часто встречающийся лейкоз в западной Европе (30% всех лейкозов, в Азии – 2-5%)
- 16. Критерии диагноза В-ХЛЛ (NCI - sponsored СLL Working group USA) Абсолютный лимфоцитоз в крови более 5х109/л
- 17. Хронический лимфолейкоз характерна слабая экспрессия SmIg (k / λ+)
- 18. Алгоритм диагностики В-ХЛЛ
- 19. ХЛЛ. Увеличение морфологически зрелых лимфоцитов в крови Абсолютный лимфоцитоз в крови 5х109/л с преобладанием малых зрелых
- 20. Гистологическая картина костного мозга при ХЛЛ
- 21. ХЛЛ. Иммуноцитохимия и иммуногистохимия
- 22. Наиболее типичные хромосомные нарушения при В- ХЛЛ Хромосомные нарушения встречаются в 50% случаев
- 23. Классификация В-ХЛЛ Rai Binet 0 Только лимфоцитоз А Лимфоцитоз + менее 3 областей поражения. I Лимфоцитозоз
- 24. Симптомы, требующие дифференциального диагноза с другими заболеваниями Лейкоцитоз Лимфоцитоз Гипогаммаглобулинемия / иммунодефицит, частые инфекции Лимфоаденопатия Увеличение
- 25. Дифференциальный диагноз ХЛЛ (1) Инфекции туберкулез Вирусные (инфекционный мононуклеоз, EBV, CMV, бруцеллез) Гиперактивная малярия со спленомегалией
- 26. Дифференциальный диагноз ХЛЛ (2) Опухолевые заболевания В-клеточные Пролимфоцитарный лейкоз Лейкемическая фаза неходжкинских лимфом - лимфома из
- 27. Дифференциальный диагноз ХЛЛ (3) Опухолевые заболевания Т-клеточные Пролимфоцитарный лейкоз Лейкемическая фаза неходжкинских лимфом - Т-клеточная лейкемия/лимфома
- 28. Выживаемость больных ХЛЛ А стадии в зависимости от стабильного и прогрессирующего течения 100% 50% 10 лет
- 29. Определение группы риска пациентов Высокий риск Низкий риск Время удвоения лимфоцитов менее 6 месяцев Время удвоения
- 30. Общая выживаемость больных ХЛЛ стадии А по Binet в зависимости от наличия соматической мутации генов, ответственных
- 31. Выживаемость больных ХЛЛ без прогрессии в зависимости от хромосомных нарушений
- 32. Выживаемость без лечения больных ХЛЛ Rai 0 cтадии в зависимости от CD38+ экспрессии M. Gentile et
- 33. Выживаемость больных ХЛЛ в зависимости от IgVH мутации и CD38 экспрессии N.Damle et al
- 34. mut JgVн(-) CD38+ выживаемость 8 лет mut JgVн(+) CD38- выживаемость 25 лет Другие сочетания 15 лет
- 35. Основные показания для начала терапии больных ХЛЛ Перспектива выполнения ТСК (молодой возраст) Прогрессирующая анемия и тромбоцитопения
- 36. Факторы, влияющие на выбор терапии больных ХЛЛ Возраст ≤ 60 лет, соматический статус?, сопутствующие заболевания? Группа
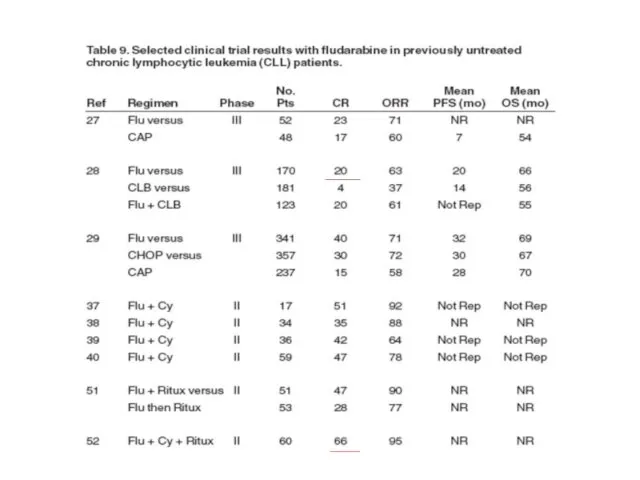
- 37. Методы лечения ХЛЛ Хлорбутин ± преднизолон СНОР, САР Флударабин (кладрибин, пентостатин) Флударабин+циклофосфамид Флударабин+ритуксимаб флударабин+циклофосфамид+ ритуксимаб Флударабин
- 38. Критерии полной ремиссии ХЛЛ Отсутствие лимфаденопатии, спленомегалии, гепатомегалии по данным КТ Отсутствие В симптомов Нормальные показатели
- 39. Роль трансплантации стволовых клеток в лечении ХЛЛ
- 41. Тактика лечения больных ХЛЛ на постремиссионном этапе После достижения ПР в группе низкого риска наблюдение В
- 42. Потенциальные мишени для патогенетического лечения ХЛЛ (N.E.Kay e.a.,2002)
- 43. Волосатоклеточный лейкоз – лимфопролиферативное заболевание, характеризующееся появлением в крови или костном мозге ≥10% характерных патологических мононуклеарных
- 44. Иммунофенотип В-клеточных лимфопролиферативных заболеваний SmIg CD5 CD22 CD23 CD25 FMC7 CD103 CD11c ХЛЛ -/+ + -/+
- 45. Лабораторные признаки при волосатоклеточном лейкозе Количество “волосатых” клеток в крови или костном мозге ≥ 10% Лейкопения
- 46. Клинические проявления при волосатоклеточном лейкозе Симптомы анемии Частые тяжелые инфекции Геморрагический синдром Выраженная спленомегалия Умеренная гепатомегалия,
- 47. Лечение волосатоклеточного лейкоза Аналоги пуриновых нуклеозидов (пентостатин, кладрибин) - 87-100% полных ремиссий, 5 летняя безрецидивная выживаемость
- 48. Лимфомы Неходжкинские лимфомы 70-88% Ходжкинские лимфомы 12-30% В мире – 4,5 млн. больных Ежегодно погибает 300000
- 49. Неходжкинские лимфомы Клинические проявления обусловлены: Наличием опухолевого очага - увеличение лимфоузлов - поражение различных органов (кожи,
- 50. Наружные лимфоузлы при неходжкинских лимфомах Эластической, затем плотной консистенции Без местных признаков воспаления Не спаяны с
- 51. Локализация первичных проявлений при неходжкинских лимфомах Периферические лимфоузлы Органы брюшной полости (диспептические проявления аппендицит симптомы кишечной
- 52. Симптомы опухолевой интоксикации Необъяснимая потливость Повышение температуры тела Похудание (≥10%) Вялость, утомляемость, снижение аппетита
- 53. Диагностический алгоритм при неходжкинских лимфомах Врачебный осмотр и сбор анамнеза Клинический анализ крови, биохимический анализ крови
- 54. Определение стадий заболевания по Энн Эрбор Стадия I Поражение одиночного регионарного лимфатического узла или одиночная локализация
- 55. МРТ. Лимфома средостения с прорастанием в ткань легкого
- 56. Магнитно-резонансная томография Лимфома легкого
- 57. Компьютерная томография. Множественное очаговое поражение легких лимфома
- 58. Лимфома желудка
- 59. Лимфома желудка эндоскопически представляется как распространенная крупнобугристая опухоль с признаками инфильрации, изъязвлениями, фибринозными наложениями
- 60. Обширная опухоль желудка, оказавшаяся лимфомой
- 61. Неходжкинская лимфома
- 62. Т-клеточная лимфома кожи
- 63. Т-клеточная лимфома кожи
- 64. Неходжкинские лимфомы низкой степени злокачественности В-клеточные Фолликулярная НХЛ (I-II степени) Диффузная лимфоцитарная НХЛ Экстранодальная В-клеточная лимфома
- 65. Неходжкинские лимфомы высокой степени злокачественности В-клеточные Диффузная крупноклеточная НХЛ НХЛ Беркитта и беркитоподобные Т-клеточные Лимфобластная лимфома/лейкемия
- 66. Лечение НХЛ Основа лечения НХЛ – химиотерапия и лучевая терапия (при некоторых формах I-IIстадиях) СОР, COAP
- 67. Грибовидный микоз является самой частой злокачественной лимфомой кожи. Начальная стадия характеризуется появлением медленно прогрессирующих "экзематоидных" пятен.
- 68. Обширное изъязвление на коже живота у больного с опухолевой стадией грибовидного микоза
- 69. Синдром Сезари - сочетание эритродермии, сопровождающейся зудом, генерализованной лимфаденопатии, а также наличие атипичных лимфоцитов (клетки Сезари)
- 70. Атипичный лимфоцит (клетка Сезари)
- 71. B-клеточные лимфомы кожи В отличие от T-клеточных лимфом, B-клеточные лимфомы, составляющие примерно 25 % от всех
- 72. Лечение лимфом кожи Стадии Ia-IIa PUVA –терапия ± α-интерферон Местная рентгенотерапия (лучевая) или химиотерапия (кармустин) Ретиноиды
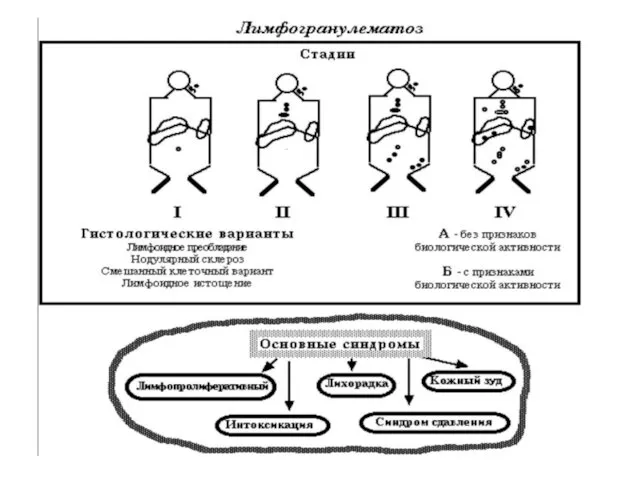
- 73. Лимфогранулематоз (болезнь Ходжкина, ходжскинская лимфома) - первичное опухолевое заболевание лимфоидной ткани. Возникает локально в одном из
- 74. Клетки Штенберга-Рида CD15+
- 75. Лимфогранулематоз, нодулярный тип лимфоидного преобладания Классическая лимфома Ходжкина, нодулярный склероз (I и II тип) Классическая лимфома
- 77. Лечение лимфогрануломатоза Химиотерапия/Лучевая терапия 36-40 Гр ( I – IIa стадии) Химиотерапия ( IIа –IVb стадии)
- 78. Схемы химиотерапии лимфогрануломатоза АВVD BEACOPP МОРР СОРР Dexa-BEAM BEAM
- 79. Эффективность лечения лимфогрануломатоза III-IVст (V. Diehl et al, 2003)
- 80. Множественная миелома Множественная миелома – лимфопролиферативное заболевание, морфологическим субстратом которого являются плазматические клетки, продуцирующие моноклональный иммуноглобулин.
- 81. По классификации REAL, миелома относится к лимфоидным опухолям низкой степени злокачественности. Множественная миелома составляет 1% онкологических
- 82. Диагностические критерии множественной миеломы ( требуется наличие всех трех) 1. Моноклональные плазматические клетки в костном мозге
- 83. Иммунофенотип миеломных клеток При множественной миеломе в костном мозге можно выделить два варианта фенотипа миеломных клеток:
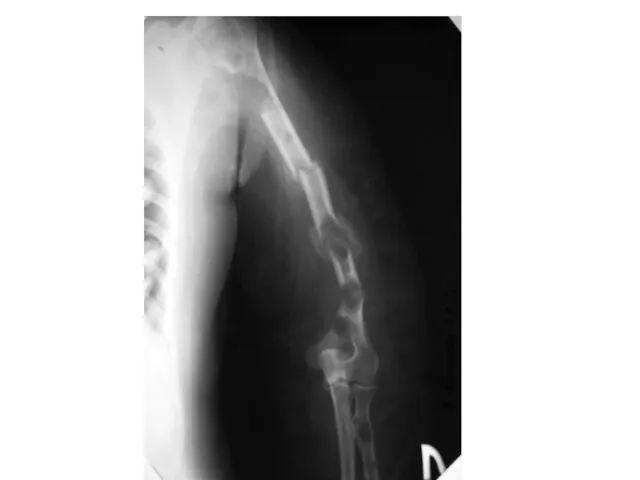
- 84. Клинические проявления миеломы Поражение костей Солитарные или множественные остеолитические повреждения Диффузный остеопороз (остеопения) Костные переломы Уменьшение
- 85. Периферическая кровь Анемия, лейкопения, тромбоцитопения, ускорение СОЭ Нарушение свертывания крови Плазмоклеточный лейкоз (плазмобласты) Циркулирующие моноклональные В-лимфоциты
- 86. Нарушения со стороны почек Протеинурия, цилиндры без эритроцитов и лейкоцитов Канальцевая дисфункция с ацидозом Почечная недостаточность
- 87. Лабораторные исследования при миеломе Полный клинический анализ крови, СОЭ Биохимическое исследование сыворотки крови с оценкой: общего
- 88. Электрофорез –иммунофиксация (сыворотки или концентрированной мочи) Количественная оценка уровня иммуноглобулинов сыворотки крови Рентгенологическое исследование костей, КТ,
- 89. Множественная миелома (IgG, IgA, IgD, IgЕ и свободные легкие цепи Каппа или Ламбда). 1. Симптоматическая миелома.
- 90. Иммунохимические варианты множественной миеломы Вариант Частота,% G-миелома 55-65 А-миелома 20-25 D-миелома 2-5 Е-миелома Точно не установлена
- 91. Стадии множественной миеломы по Durie/Salmon. Стадия Критерии 1 А,В Уровень Hb >100 г/л; Нормальный уровень кальция
- 92. Прогностические факторы при множественной миеломе Возраст Соматический статус Β2-микроглобулин Альбумин сыворотки Креатинин сыворотки Лактатдегидрогеназа СРБ Уровень
- 93. Моноклональный образец М-протеина сыворотки при денситометрии после электрофореза на агарозном геле. Пик в гамма-фракции.
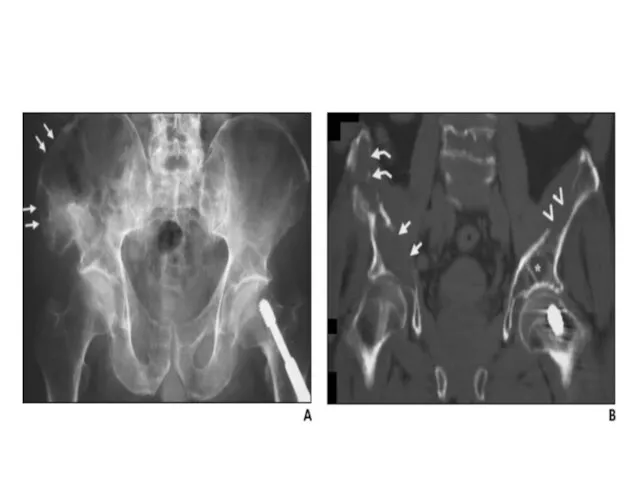
- 94. А- рентгенограмма, В- мультидетекторная КТ (МДКТ), С- магнитнорезонансная томография (МР)
- 97. Плазматические клетки в костном мозге больного множественной миеломой
- 99. Кортикостероиды Пульс дексаметазона VAD Повторяется каждые 3-4 нед Декса 40 мг ро день 1-4, 9-12, 17-20
- 100. Bortezomid (PS-341, Велкейд) 1,3 мг/м2в день ро дни 1,4,8,11 каждые 21день - 25 Триоксид мышьяка (
- 101. Лечение осложнений миеломы Осложнение Терапевтические подходы Поражение костей Бифосфонаты (бонефос, аредиа, зомета) поощрение двигательной активности для
- 102. Анемия при симптомах анемии эритропоэтин во время химиотерапии При необходимости - гемотрансфузии Инфекции Назначение антибиотиков широкого
- 103. Почечная недостаточ-ность Коррекция обратимых причин, таких как дегидратация, гиперкальциемия, гиперурикемии Химиотерапия для быстрого контроля за заболеванием
- 104. Макроглобулинемия Вальденстрема. Характеризуется наличием моноклонального иммуноглобулина М (PIgM), высокой вязкостью сыворотки крови и геморрагическим синдромом без
- 105. Клиническая манифестация макроглобулинемии Вальденстрема Симптомы Признаки Слабость, усталость Симптомы гипервязкости Ночная потливость Потеря веса Периферическая нейропатия
- 106. Признаки синдрома гипервязкости Кровотечение из носа и десен, реже – желудочно-кишечные и послеоперационные) Пурпура Нарушения зрения
- 108. Скачать презентацию
Общая схема кроветворения
Общая схема кроветворения

Этапы созревания лимфоцитов
костный мозг
стволовая клетка – клетка предшественница
В-лимфоцит Т-лимфоцит
лимфоузлы
Этапы созревания лимфоцитов
костный мозг
стволовая клетка – клетка предшественница
В-лимфоцит Т-лимфоцит
лимфоузлы

Этиопатогенез лимфопролиферативных заболеваний
Предрасполагающие факторы:
хроническая антигенная стимуляция,
иммунодефицит и вирусные инфекции.
Первичное
Этиопатогенез лимфопролиферативных заболеваний
Предрасполагающие факторы:
хроническая антигенная стимуляция,
иммунодефицит и вирусные инфекции.
Первичное

Генетические повреждения при лимфомах можно подразделить на две крупные категории: активация
Генетические повреждения при лимфомах можно подразделить на две крупные категории: активация
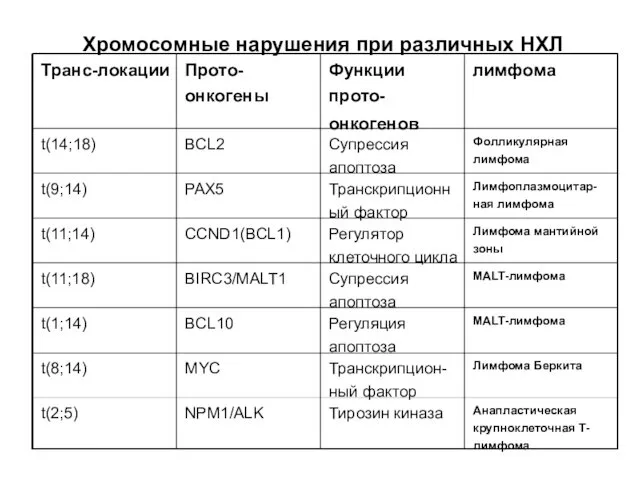
Хромосомные нарушения при различных НХЛ
Транс-локации
Прото-онкогены
Функции прото-
онкогенов
лимфома
t(14;18)
BCL2
Супрессия апоптоза
Фолликулярная лимфома
t(9;14)
PAX5
Транскрипционный фактор
Лимфоплазмоцитар-ная лимфома
t(11;14)
CCND1(BCL1)
Регулятор клеточного
Хромосомные нарушения при различных НХЛ
Транс-локации
Прото-онкогены
Функции прото-
онкогенов
лимфома
t(14;18)
BCL2
Супрессия апоптоза
Фолликулярная лимфома
t(9;14)
PAX5
Транскрипционный фактор
Лимфоплазмоцитар-ная лимфома
t(11;14)
CCND1(BCL1)
Регулятор клеточного

При созревании от костно-мозговой клетки-предшественницы лимфопоэза до плазматической клетки, геном В-лимфоцитов
При созревании от костно-мозговой клетки-предшественницы лимфопоэза до плазматической клетки, геном В-лимфоцитов
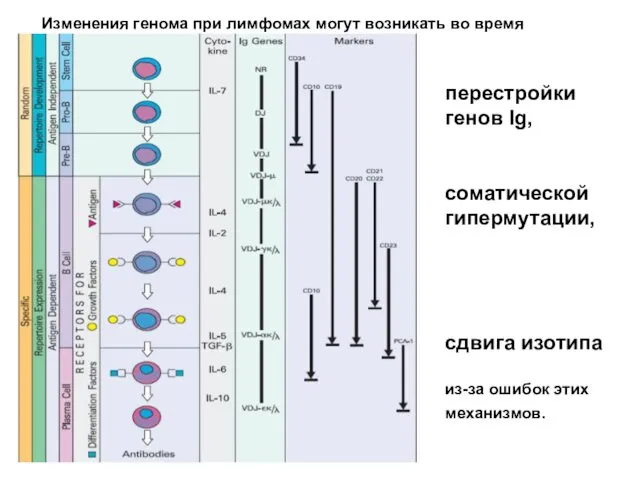
перестройки генов Ig,
соматической гипермутации,
сдвига изотипа
из-за ошибок этих механизмов.
Изменения
перестройки генов Ig,
соматической гипермутации,
сдвига изотипа
из-за ошибок этих механизмов.
Изменения

Главная особенность лимфомагенеза в преобладании среди хромосомных нарушений транслокаций с участием
Главная особенность лимфомагенеза в преобладании среди хромосомных нарушений транслокаций с участием

В-клеточные опухоли с фенотипом зрелых лимфоцитов
Хронический лимфоцитарный лейкоз / лимфоцитарная
В-клеточные опухоли с фенотипом зрелых лимфоцитов
Хронический лимфоцитарный лейкоз / лимфоцитарная

Классификация ВОЗ
лимфопролиферативных заболеваний (2)
В-клеточные лимфопролиферативные процессы с неопределенным опухолевым потенциалом
Классификация ВОЗ
лимфопролиферативных заболеваний (2)
В-клеточные лимфопролиферативные процессы с неопределенным опухолевым потенциалом

В-клеточные лимфопролиферативные заболевания, не вошедшие в классификацию ВОЗ
Внутрисосудистая
В-крупноклеточная лимфома
Первичный амилоидоз
В-клеточные лимфопролиферативные заболевания, не вошедшие в классификацию ВОЗ
Внутрисосудистая
В-крупноклеточная лимфома
Первичный амилоидоз

T- и NK-клеточные опухоли с фенотипом зрелых лимфоцитов
Лейкозы и первично
T- и NK-клеточные опухоли с фенотипом зрелых лимфоцитов
Лейкозы и первично

В-клеточный хронический лимфолейкоз - моноклональная пролиферация CD5-позитивных зрелых В-лимфоцитов с первичным
В-клеточный хронический лимфолейкоз - моноклональная пролиферация CD5-позитивных зрелых В-лимфоцитов с первичным

Хронический лимфолейкоз
Наиболее часто встречающийся лейкоз в западной Европе (30% всех лейкозов,
Хронический лимфолейкоз
Наиболее часто встречающийся лейкоз в западной Европе (30% всех лейкозов,

Критерии диагноза В-ХЛЛ (NCI - sponsored СLL Working group USA)
Абсолютный
Критерии диагноза В-ХЛЛ (NCI - sponsored СLL Working group USA)
Абсолютный

Хронический лимфолейкоз
характерна слабая экспрессия
SmIg (k / λ+)
Хронический лимфолейкоз
характерна слабая экспрессия
SmIg (k / λ+)

Алгоритм диагностики В-ХЛЛ
Алгоритм диагностики В-ХЛЛ
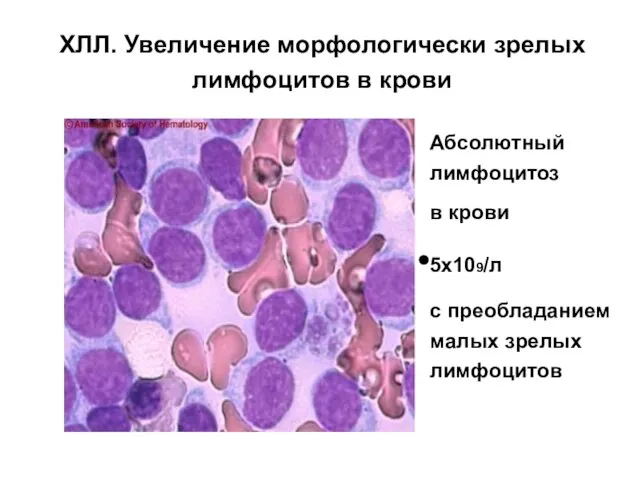
ХЛЛ. Увеличение морфологически зрелых лимфоцитов в крови
Абсолютный лимфоцитоз
в крови
5х109/л
ХЛЛ. Увеличение морфологически зрелых лимфоцитов в крови
Абсолютный лимфоцитоз
в крови
5х109/л
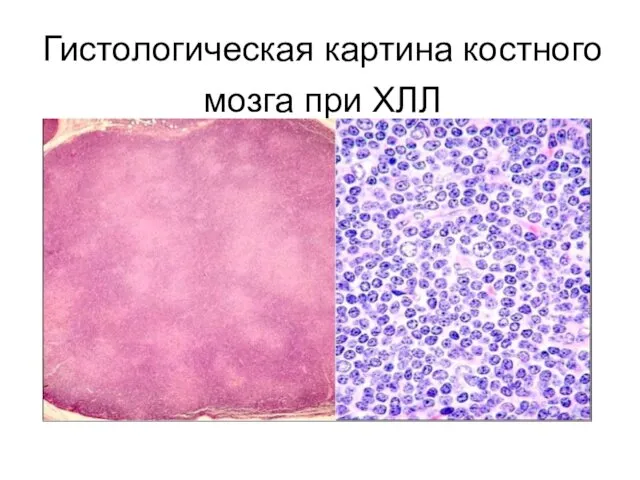
Гистологическая картина костного мозга при ХЛЛ
Гистологическая картина костного мозга при ХЛЛ
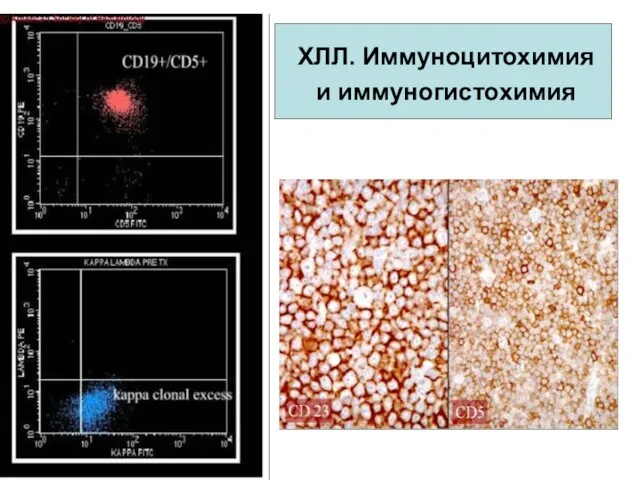
ХЛЛ. Иммуноцитохимия
и иммуногистохимия
ХЛЛ. Иммуноцитохимия
и иммуногистохимия

Наиболее типичные хромосомные нарушения при В- ХЛЛ
Хромосомные нарушения встречаются в 50%
Наиболее типичные хромосомные нарушения при В- ХЛЛ
Хромосомные нарушения встречаются в 50%
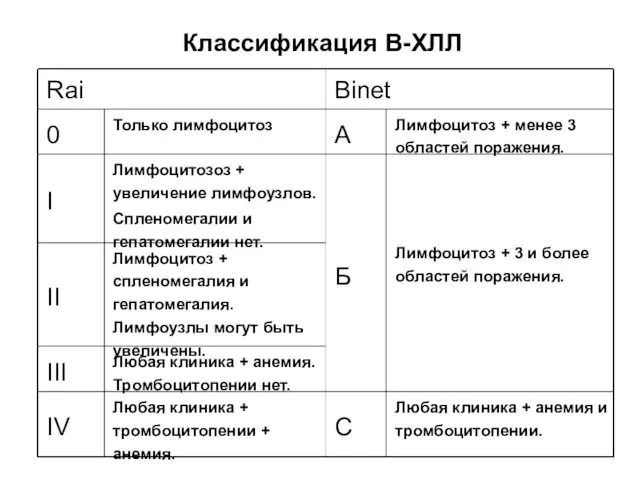
Классификация В-ХЛЛ
Rai
Binet
0
Только лимфоцитоз
А
Лимфоцитоз + менее 3 областей поражения.
I
Лимфоцитозоз + увеличение
Классификация В-ХЛЛ
Rai
Binet
0
Только лимфоцитоз
А
Лимфоцитоз + менее 3 областей поражения.
I
Лимфоцитозоз + увеличение

Симптомы, требующие дифференциального диагноза с другими заболеваниями
Лейкоцитоз
Лимфоцитоз
Гипогаммаглобулинемия / иммунодефицит, частые
Симптомы, требующие дифференциального диагноза с другими заболеваниями
Лейкоцитоз
Лимфоцитоз
Гипогаммаглобулинемия / иммунодефицит, частые

Дифференциальный диагноз ХЛЛ (1)
Инфекции
туберкулез
Вирусные (инфекционный мононуклеоз, EBV, CMV, бруцеллез)
Гиперактивная малярия со
Дифференциальный диагноз ХЛЛ (1)
Инфекции
туберкулез
Вирусные (инфекционный мононуклеоз, EBV, CMV, бруцеллез)
Гиперактивная малярия со

Дифференциальный диагноз ХЛЛ (2)
Опухолевые заболевания В-клеточные
Пролимфоцитарный лейкоз
Лейкемическая фаза неходжкинских лимфом
- лимфома
Дифференциальный диагноз ХЛЛ (2)
Опухолевые заболевания В-клеточные
Пролимфоцитарный лейкоз
Лейкемическая фаза неходжкинских лимфом
- лимфома

Дифференциальный диагноз ХЛЛ (3)
Опухолевые заболевания Т-клеточные
Пролимфоцитарный лейкоз
Лейкемическая фаза неходжкинских лимфом
- Т-клеточная
Дифференциальный диагноз ХЛЛ (3)
Опухолевые заболевания Т-клеточные
Пролимфоцитарный лейкоз
Лейкемическая фаза неходжкинских лимфом
- Т-клеточная
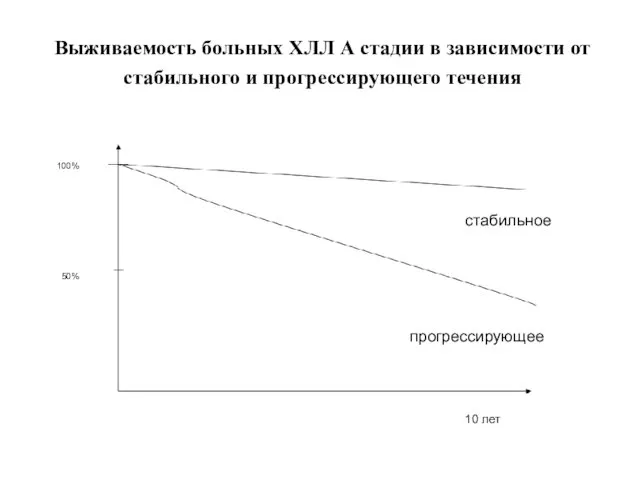
Выживаемость больных ХЛЛ А стадии в зависимости от стабильного и прогрессирующего
Выживаемость больных ХЛЛ А стадии в зависимости от стабильного и прогрессирующего
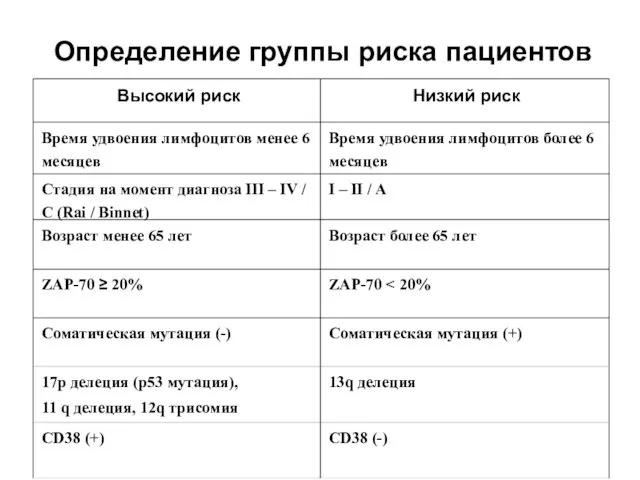
Определение группы риска пациентов
Высокий риск
Низкий риск
Время удвоения лимфоцитов менее 6 месяцев
Время
Определение группы риска пациентов
Высокий риск
Низкий риск
Время удвоения лимфоцитов менее 6 месяцев
Время
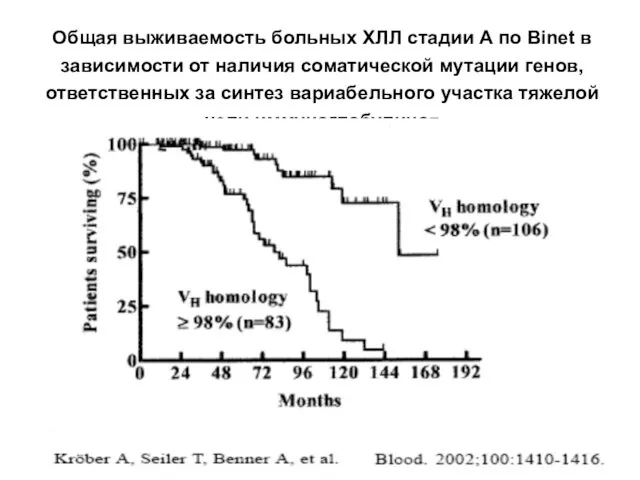
Общая выживаемость больных ХЛЛ стадии А по Binet в зависимости от
Общая выживаемость больных ХЛЛ стадии А по Binet в зависимости от
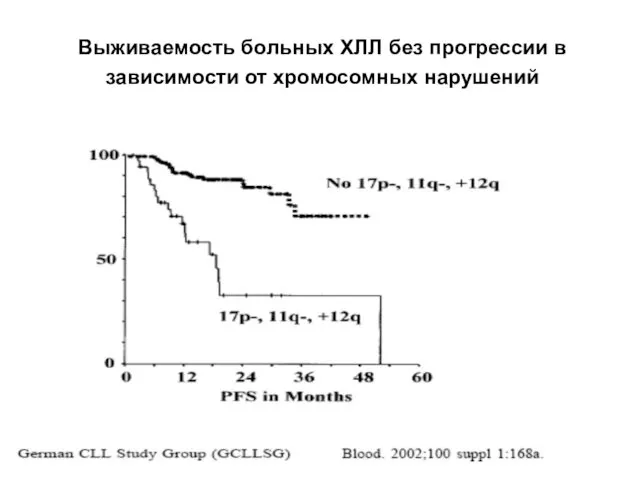
Выживаемость больных ХЛЛ без прогрессии в зависимости от хромосомных нарушений
Выживаемость больных ХЛЛ без прогрессии в зависимости от хромосомных нарушений
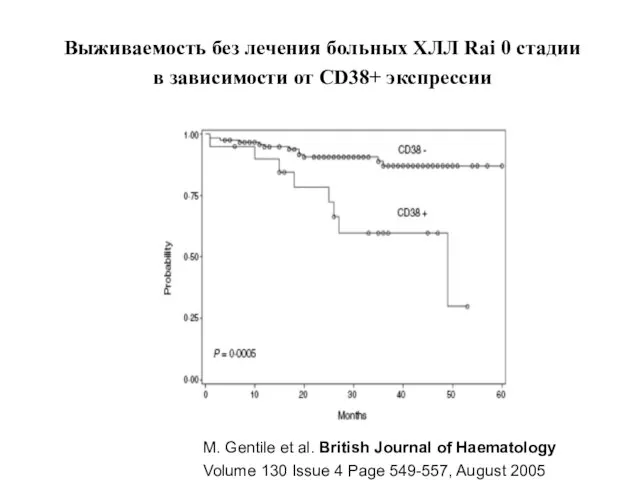
Выживаемость без лечения больных ХЛЛ Rai 0 cтадии
в зависимости от
Выживаемость без лечения больных ХЛЛ Rai 0 cтадии в зависимости от
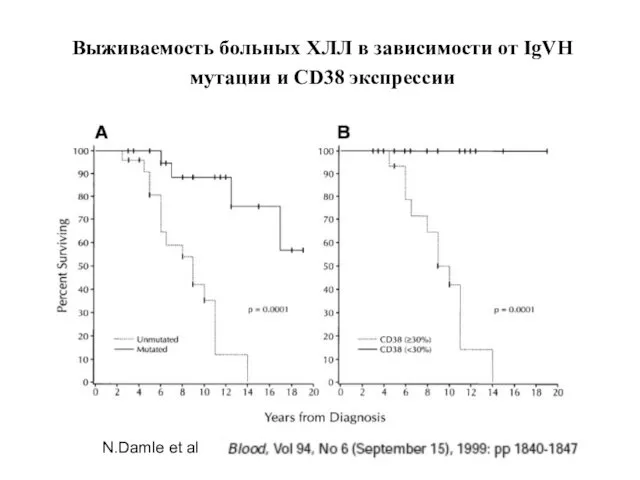
Выживаемость больных ХЛЛ в зависимости от IgVH мутации и CD38 экспрессии
N.Damle
Выживаемость больных ХЛЛ в зависимости от IgVH мутации и CD38 экспрессии
N.Damle

mut JgVн(-) CD38+ выживаемость 8 лет
mut JgVн(+) CD38- выживаемость 25
mut JgVн(-) CD38+ выживаемость 8 лет
mut JgVн(+) CD38- выживаемость 25

Основные показания для начала терапии больных ХЛЛ
Перспектива выполнения ТСК (молодой возраст)
Основные показания для начала терапии больных ХЛЛ
Перспектива выполнения ТСК (молодой возраст)

Факторы, влияющие на выбор терапии больных ХЛЛ
Возраст ≤ 60 лет, соматический
Факторы, влияющие на выбор терапии больных ХЛЛ
Возраст ≤ 60 лет, соматический

Методы лечения ХЛЛ
Хлорбутин ± преднизолон
СНОР, САР
Флударабин (кладрибин, пентостатин)
Флударабин+циклофосфамид
Флударабин+ритуксимаб
флударабин+циклофосфамид+
ритуксимаб
Флударабин + циклофосфамид +
Методы лечения ХЛЛ
Хлорбутин ± преднизолон
СНОР, САР
Флударабин (кладрибин, пентостатин)
Флударабин+циклофосфамид
Флударабин+ритуксимаб
флударабин+циклофосфамид+
ритуксимаб
Флударабин + циклофосфамид +

Критерии полной ремиссии ХЛЛ
Отсутствие лимфаденопатии, спленомегалии, гепатомегалии по данным КТ
Отсутствие В
Критерии полной ремиссии ХЛЛ
Отсутствие лимфаденопатии, спленомегалии, гепатомегалии по данным КТ
Отсутствие В
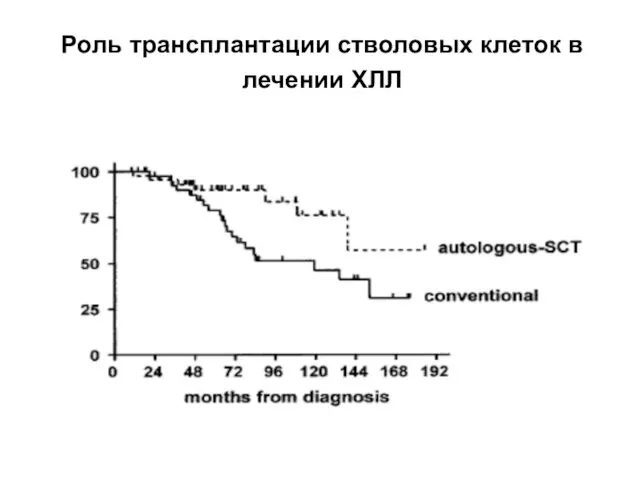
Роль трансплантации стволовых клеток в лечении ХЛЛ
Роль трансплантации стволовых клеток в лечении ХЛЛ

Тактика лечения больных ХЛЛ на постремиссионном этапе
После достижения ПР в группе
Тактика лечения больных ХЛЛ на постремиссионном этапе
После достижения ПР в группе

Потенциальные мишени для патогенетического лечения ХЛЛ (N.E.Kay e.a.,2002)
Потенциальные мишени для патогенетического лечения ХЛЛ (N.E.Kay e.a.,2002)

Волосатоклеточный лейкоз – лимфопролиферативное заболевание, характеризующееся появлением в крови или костном
Волосатоклеточный лейкоз – лимфопролиферативное заболевание, характеризующееся появлением в крови или костном
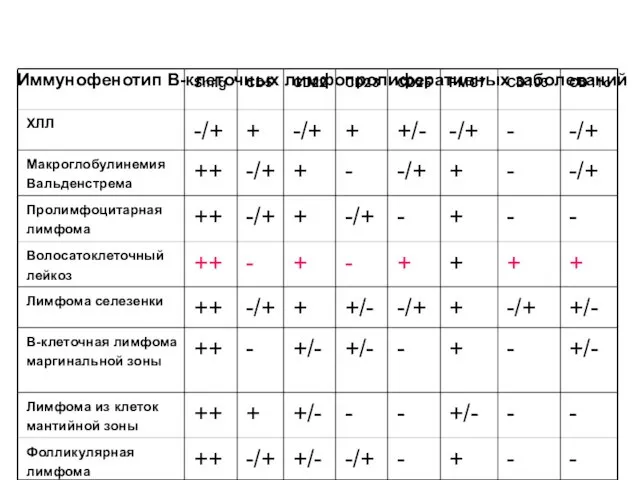
Иммунофенотип В-клеточных лимфопролиферативных заболеваний
SmIg
CD5
CD22
CD23
CD25
FMC7
CD103
CD11c
ХЛЛ
-/+
+
-/+
+
+/-
-/+
-
-/+
Макроглобулинемия Вальденстрема
++
-/+
+
-
-/+
+
-
-/+
Пролимфоцитарная лимфома
++
-/+
+
-/+
-
+
-
-
Волосатоклеточный лейкоз
++
-
+
-
+
+
+
+
Лимфома селезенки
++
-/+
+
+/-
-/+
+
-/+
+/-
В-клеточная лимфома маргинальной
Иммунофенотип В-клеточных лимфопролиферативных заболеваний
SmIg
CD5
CD22
CD23
CD25
FMC7
CD103
CD11c
ХЛЛ
-/+
+
-/+
+
+/-
-/+
-
-/+
Макроглобулинемия Вальденстрема
++
-/+
+
-
-/+
+
-
-/+
Пролимфоцитарная лимфома
++
-/+
+
-/+
-
+
-
-
Волосатоклеточный лейкоз
++
-
+
-
+
+
+
+
Лимфома селезенки
++
-/+
+
+/-
-/+
+
-/+
+/-
В-клеточная лимфома маргинальной

Лабораторные признаки при волосатоклеточном лейкозе
Количество “волосатых” клеток в крови или костном
Лабораторные признаки при волосатоклеточном лейкозе
Количество “волосатых” клеток в крови или костном

Клинические проявления при волосатоклеточном лейкозе
Симптомы анемии
Частые тяжелые инфекции
Геморрагический синдром
Выраженная спленомегалия
Умеренная гепатомегалия,
Клинические проявления при волосатоклеточном лейкозе
Симптомы анемии
Частые тяжелые инфекции
Геморрагический синдром
Выраженная спленомегалия
Умеренная гепатомегалия,

Лечение волосатоклеточного лейкоза
Аналоги пуриновых нуклеозидов (пентостатин, кладрибин) - 87-100% полных ремиссий,
Лечение волосатоклеточного лейкоза
Аналоги пуриновых нуклеозидов (пентостатин, кладрибин) - 87-100% полных ремиссий,

Лимфомы
Неходжкинские лимфомы 70-88%
Ходжкинские лимфомы 12-30%
В мире – 4,5 млн. больных
Ежегодно
Лимфомы
Неходжкинские лимфомы 70-88%
Ходжкинские лимфомы 12-30%
В мире – 4,5 млн. больных
Ежегодно

Неходжкинские лимфомы
Клинические проявления обусловлены:
Наличием опухолевого очага
- увеличение лимфоузлов
- поражение различных органов
Неходжкинские лимфомы
Клинические проявления обусловлены:
Наличием опухолевого очага
- увеличение лимфоузлов
- поражение различных органов

Наружные лимфоузлы при неходжкинских лимфомах
Эластической, затем плотной консистенции
Без местных признаков воспаления
Не
Наружные лимфоузлы при неходжкинских лимфомах
Эластической, затем плотной консистенции
Без местных признаков воспаления
Не

Локализация первичных проявлений при неходжкинских лимфомах
Периферические лимфоузлы
Органы брюшной полости
(диспептические проявления
аппендицит
симптомы кишечной
Локализация первичных проявлений при неходжкинских лимфомах
Периферические лимфоузлы
Органы брюшной полости
(диспептические проявления
аппендицит
симптомы кишечной

Симптомы опухолевой интоксикации
Необъяснимая потливость
Повышение температуры тела
Похудание (≥10%)
Вялость, утомляемость, снижение аппетита
Симптомы опухолевой интоксикации
Необъяснимая потливость
Повышение температуры тела
Похудание (≥10%)
Вялость, утомляемость, снижение аппетита

Диагностический алгоритм при неходжкинских лимфомах
Врачебный осмотр и сбор анамнеза
Клинический анализ крови,
Диагностический алгоритм при неходжкинских лимфомах
Врачебный осмотр и сбор анамнеза
Клинический анализ крови,

Определение стадий заболевания по Энн Эрбор
Стадия I
Поражение одиночного регионарного
Определение стадий заболевания по Энн Эрбор
Стадия I Поражение одиночного регионарного
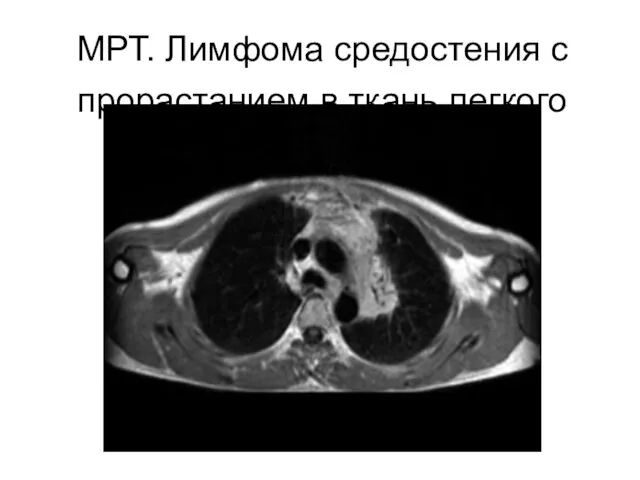
МРТ. Лимфома средостения с прорастанием в ткань легкого
МРТ. Лимфома средостения с прорастанием в ткань легкого
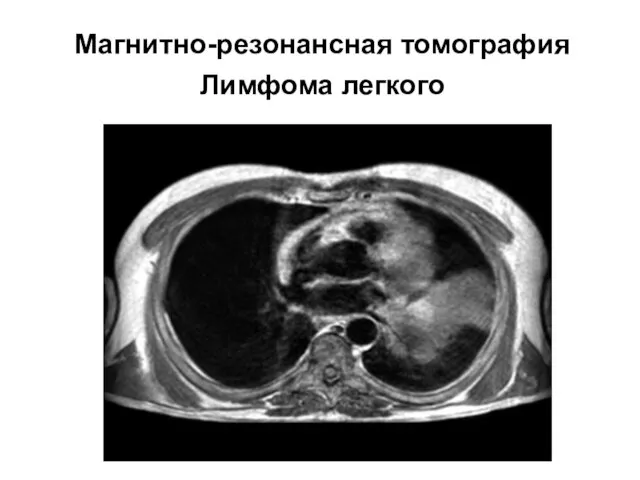
Магнитно-резонансная томография
Лимфома легкого
Магнитно-резонансная томография
Лимфома легкого
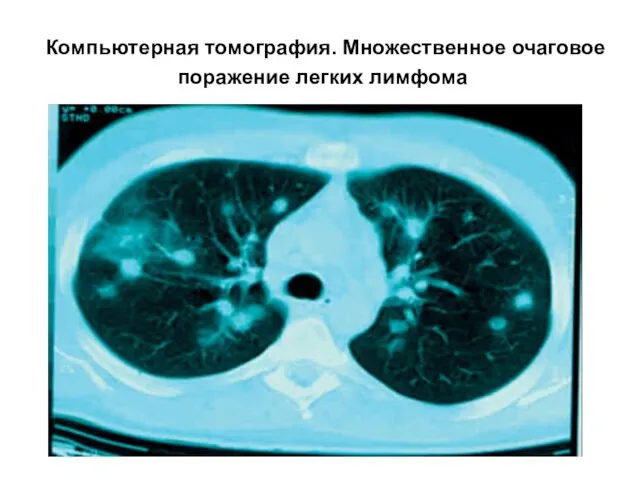
Компьютерная томография. Множественное очаговое поражение легких лимфома
Компьютерная томография. Множественное очаговое поражение легких лимфома
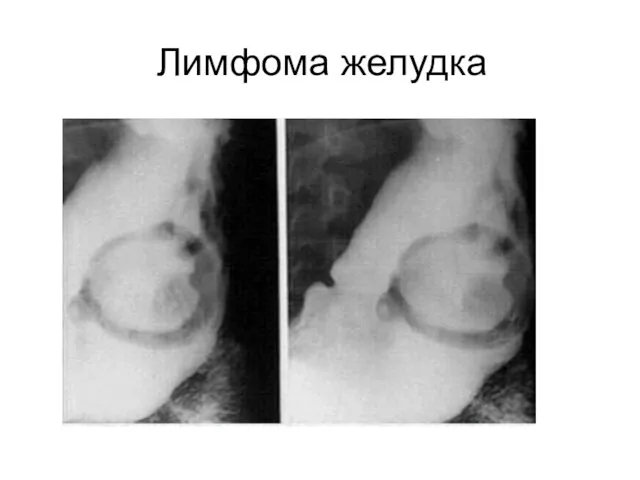
Лимфома желудка
Лимфома желудка
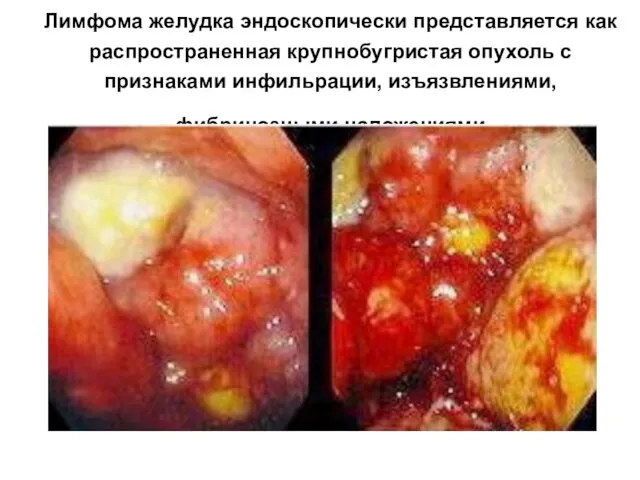
Лимфома желудка эндоскопически представляется как распространенная крупнобугристая опухоль с признаками инфильрации,
Лимфома желудка эндоскопически представляется как распространенная крупнобугристая опухоль с признаками инфильрации,
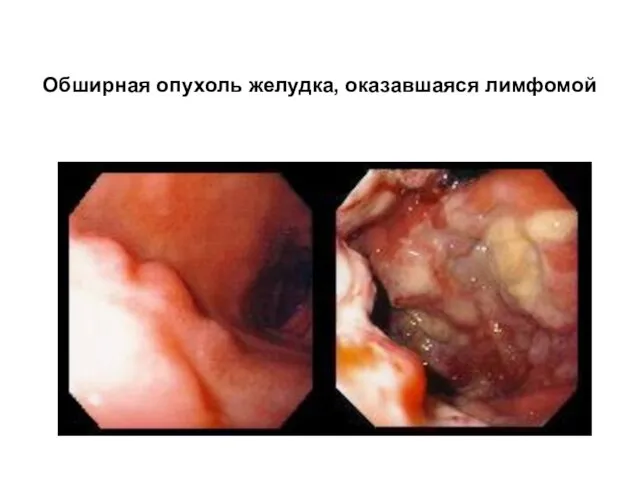
Обширная опухоль желудка, оказавшаяся лимфомой
Обширная опухоль желудка, оказавшаяся лимфомой
Неходжкинская лимфома
Неходжкинская лимфома
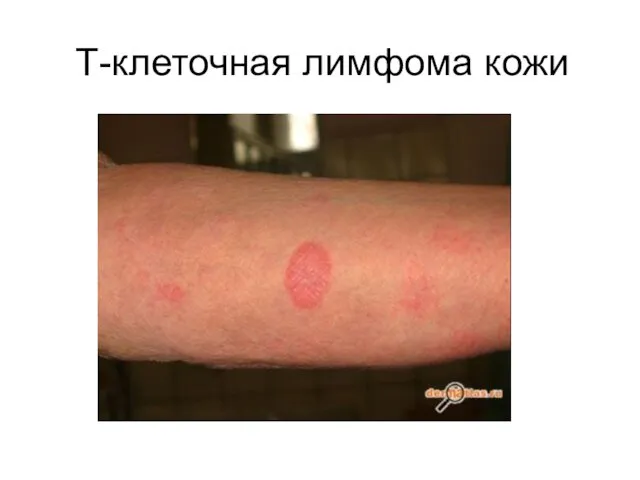
Т-клеточная лимфома кожи
Т-клеточная лимфома кожи
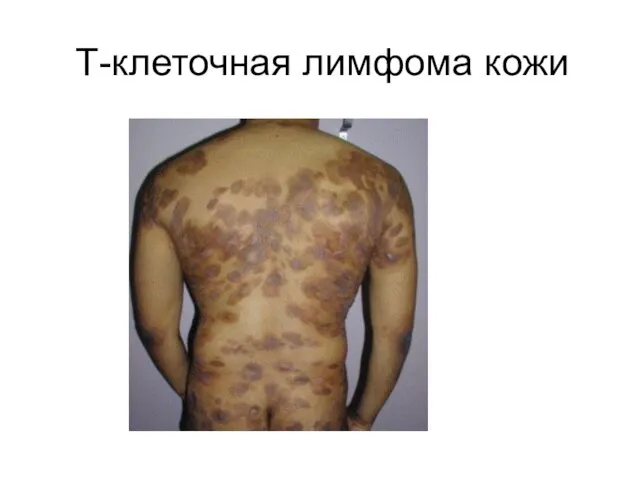
Т-клеточная лимфома кожи
Т-клеточная лимфома кожи

Неходжкинские лимфомы низкой степени злокачественности
В-клеточные
Фолликулярная НХЛ (I-II степени)
Диффузная лимфоцитарная НХЛ
Экстранодальная В-клеточная
Неходжкинские лимфомы низкой степени злокачественности
В-клеточные
Фолликулярная НХЛ (I-II степени)
Диффузная лимфоцитарная НХЛ
Экстранодальная В-клеточная

Неходжкинские лимфомы высокой степени злокачественности
В-клеточные
Диффузная крупноклеточная НХЛ
НХЛ Беркитта и беркитоподобные
Т-клеточные
Лимфобластная лимфома/лейкемия
Периферические
Неходжкинские лимфомы высокой степени злокачественности
В-клеточные
Диффузная крупноклеточная НХЛ
НХЛ Беркитта и беркитоподобные
Т-клеточные
Лимфобластная лимфома/лейкемия
Периферические

Лечение НХЛ
Основа лечения НХЛ – химиотерапия и лучевая терапия (при некоторых
Лечение НХЛ
Основа лечения НХЛ – химиотерапия и лучевая терапия (при некоторых

Грибовидный микоз является самой частой злокачественной лимфомой кожи.
Начальная стадия характеризуется
Грибовидный микоз является самой частой злокачественной лимфомой кожи.
Начальная стадия характеризуется
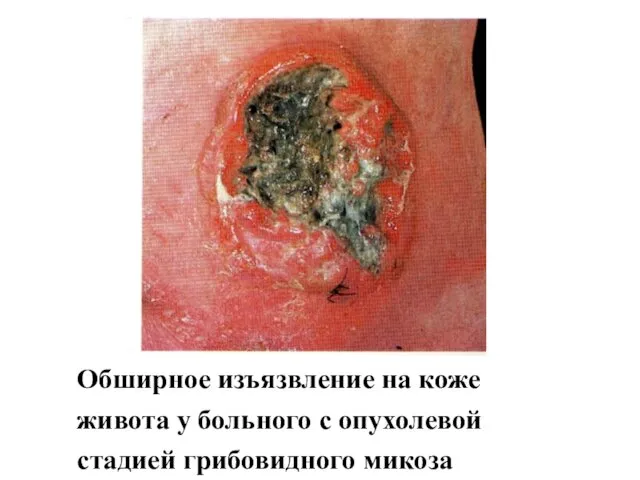
Обширное изъязвление на коже живота у больного с опухолевой стадией грибовидного
Обширное изъязвление на коже живота у больного с опухолевой стадией грибовидного

Синдром Сезари - сочетание эритродермии, сопровождающейся зудом, генерализованной лимфаденопатии, а также
Синдром Сезари - сочетание эритродермии, сопровождающейся зудом, генерализованной лимфаденопатии, а также
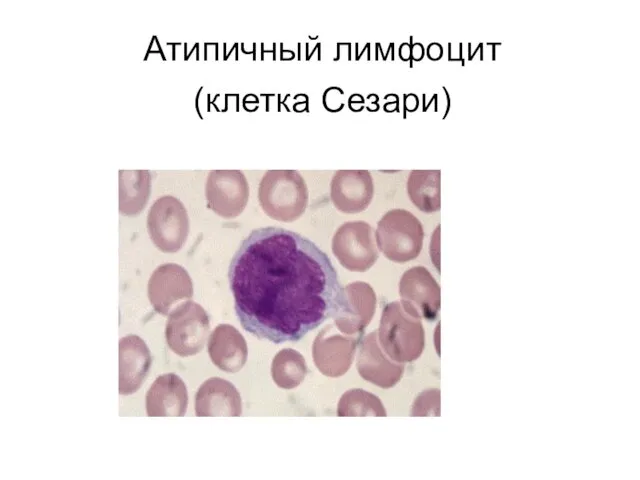
Атипичный лимфоцит
(клетка Сезари)
Атипичный лимфоцит
(клетка Сезари)

B-клеточные лимфомы кожи
В отличие от T-клеточных лимфом, B-клеточные лимфомы, составляющие примерно
B-клеточные лимфомы кожи
В отличие от T-клеточных лимфом, B-клеточные лимфомы, составляющие примерно

Лечение лимфом кожи
Стадии Ia-IIa
PUVA –терапия ± α-интерферон
Местная рентгенотерапия (лучевая) или
Лечение лимфом кожи
Стадии Ia-IIa
PUVA –терапия ± α-интерферон
Местная рентгенотерапия (лучевая) или

Лимфогранулематоз (болезнь Ходжкина, ходжскинская лимфома) - первичное опухолевое заболевание лимфоидной ткани.
Лимфогранулематоз (болезнь Ходжкина, ходжскинская лимфома) - первичное опухолевое заболевание лимфоидной ткани.
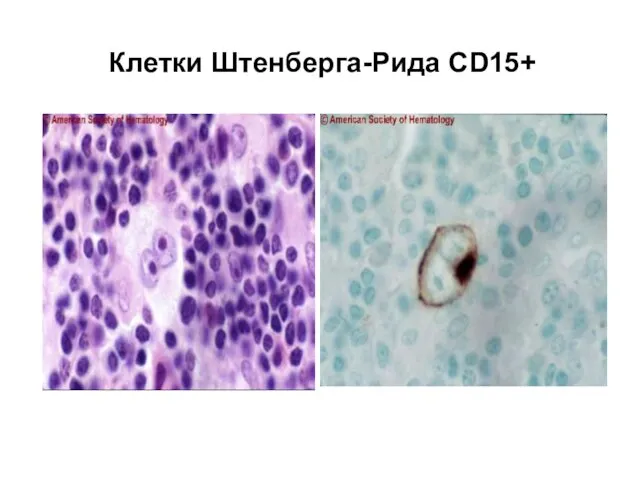
Клетки Штенберга-Рида CD15+
Клетки Штенберга-Рида CD15+

Лимфогранулематоз, нодулярный тип лимфоидного преобладания
Классическая лимфома Ходжкина, нодулярный склероз (I
Лимфогранулематоз, нодулярный тип лимфоидного преобладания
Классическая лимфома Ходжкина, нодулярный склероз (I

Лечение лимфогрануломатоза
Химиотерапия/Лучевая терапия 36-40 Гр ( I – IIa стадии)
Химиотерапия
Лечение лимфогрануломатоза
Химиотерапия/Лучевая терапия 36-40 Гр ( I – IIa стадии)
Химиотерапия

Схемы химиотерапии лимфогрануломатоза
АВVD
BEACOPP
МОРР
СОРР
Dexa-BEAM
BEAM
Схемы химиотерапии лимфогрануломатоза
АВVD
BEACOPP
МОРР
СОРР
Dexa-BEAM
BEAM
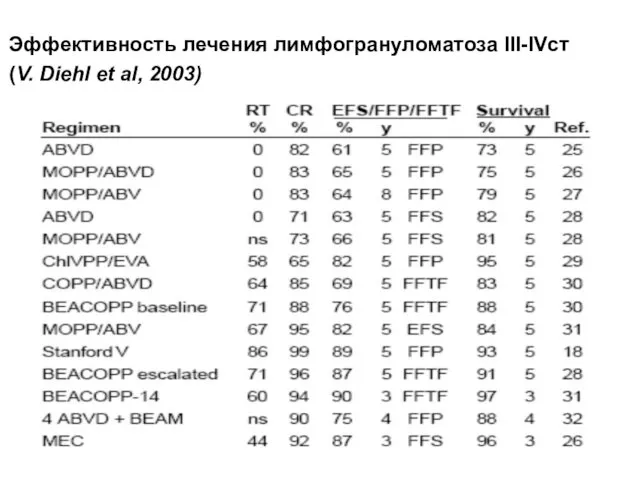
Эффективность лечения лимфогрануломатоза III-IVст
(V. Diehl et al, 2003)
Эффективность лечения лимфогрануломатоза III-IVст
(V. Diehl et al, 2003)

Множественная миелома
Множественная миелома – лимфопролиферативное заболевание, морфологическим субстратом которого являются плазматические
Множественная миелома
Множественная миелома – лимфопролиферативное заболевание, морфологическим субстратом которого являются плазматические

По классификации REAL, миелома относится к лимфоидным опухолям низкой степени злокачественности.
Множественная
По классификации REAL, миелома относится к лимфоидным опухолям низкой степени злокачественности.
Множественная

Диагностические критерии множественной миеломы ( требуется наличие всех трех)
1. Моноклональные плазматические
Диагностические критерии множественной миеломы ( требуется наличие всех трех)
1. Моноклональные плазматические

Иммунофенотип миеломных клеток
При множественной миеломе в костном мозге можно выделить два
Иммунофенотип миеломных клеток
При множественной миеломе в костном мозге можно выделить два
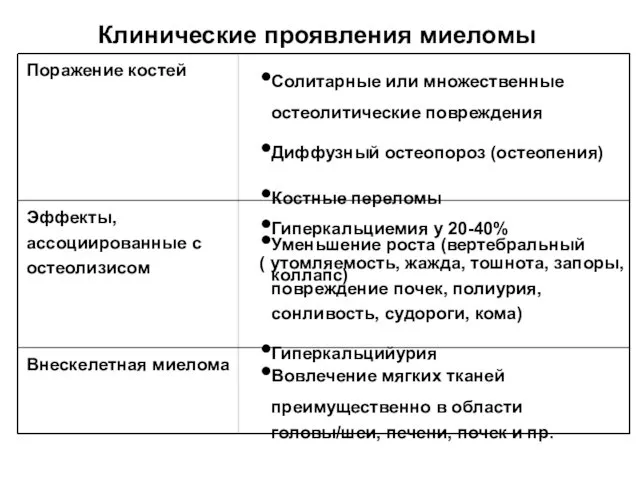
Клинические проявления миеломы
Поражение костей
Солитарные или множественные остеолитические повреждения
Диффузный остеопороз (остеопения)
Костные
Клинические проявления миеломы
Поражение костей
Солитарные или множественные остеолитические повреждения
Диффузный остеопороз (остеопения)
Костные

Периферическая кровь
Анемия, лейкопения, тромбоцитопения, ускорение СОЭ
Нарушение свертывания крови
Плазмоклеточный лейкоз (плазмобласты)
Циркулирующие
Периферическая кровь
Анемия, лейкопения, тромбоцитопения, ускорение СОЭ
Нарушение свертывания крови
Плазмоклеточный лейкоз (плазмобласты)
Циркулирующие

Нарушения со стороны почек
Протеинурия, цилиндры без эритроцитов и лейкоцитов
Канальцевая дисфункция с
Нарушения со стороны почек
Протеинурия, цилиндры без эритроцитов и лейкоцитов
Канальцевая дисфункция с

Лабораторные исследования при миеломе
Полный клинический анализ крови, СОЭ
Биохимическое исследование сыворотки
Лабораторные исследования при миеломе
Полный клинический анализ крови, СОЭ
Биохимическое исследование сыворотки

Электрофорез –иммунофиксация (сыворотки или концентрированной мочи)
Количественная оценка уровня иммуноглобулинов сыворотки крови
Рентгенологическое
Электрофорез –иммунофиксация (сыворотки или концентрированной мочи)
Количественная оценка уровня иммуноглобулинов сыворотки крови
Рентгенологическое

Множественная миелома (IgG, IgA, IgD, IgЕ и свободные легкие цепи Каппа
Множественная миелома (IgG, IgA, IgD, IgЕ и свободные легкие цепи Каппа
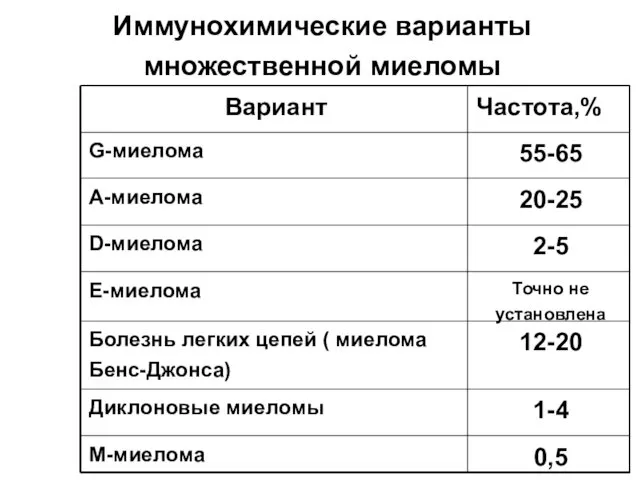
Иммунохимические варианты множественной миеломы
Вариант
Частота,%
G-миелома
55-65
А-миелома
20-25
D-миелома
2-5
Е-миелома
Точно не установлена
Болезнь легких цепей ( миелома Бенс-Джонса)
12-20
Диклоновые
Иммунохимические варианты множественной миеломы
Вариант
Частота,%
G-миелома
55-65
А-миелома
20-25
D-миелома
2-5
Е-миелома
Точно не установлена
Болезнь легких цепей ( миелома Бенс-Джонса)
12-20
Диклоновые

Стадии множественной миеломы по Durie/Salmon.
Стадия
Критерии
1
А,В
Уровень Hb >100 г/л;
Нормальный уровень кальция сыворотки;
На
Стадии множественной миеломы по Durie/Salmon.
Стадия
Критерии
1
А,В
Уровень Hb >100 г/л;
Нормальный уровень кальция сыворотки;
На

Прогностические факторы при множественной миеломе
Возраст
Соматический статус
Β2-микроглобулин
Альбумин сыворотки
Креатинин сыворотки
Лактатдегидрогеназа
СРБ
Уровень гемоглобина в крови
Количество
Прогностические факторы при множественной миеломе
Возраст
Соматический статус
Β2-микроглобулин
Альбумин сыворотки
Креатинин сыворотки
Лактатдегидрогеназа
СРБ
Уровень гемоглобина в крови
Количество
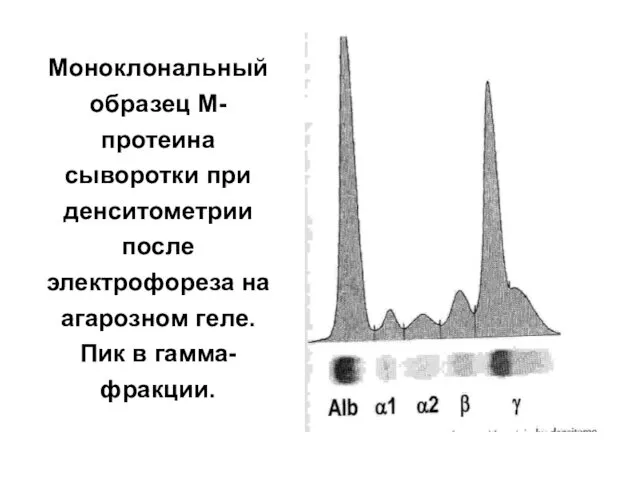
Моноклональный образец М-протеина сыворотки при денситометрии после электрофореза на агарозном геле.
Моноклональный образец М-протеина сыворотки при денситометрии после электрофореза на агарозном геле.
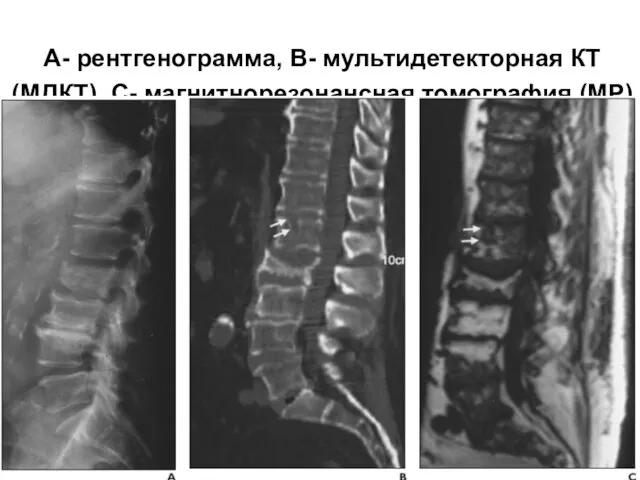
А- рентгенограмма, В- мультидетекторная КТ (МДКТ), С- магнитнорезонансная томография (МР)
А- рентгенограмма, В- мультидетекторная КТ (МДКТ), С- магнитнорезонансная томография (МР)
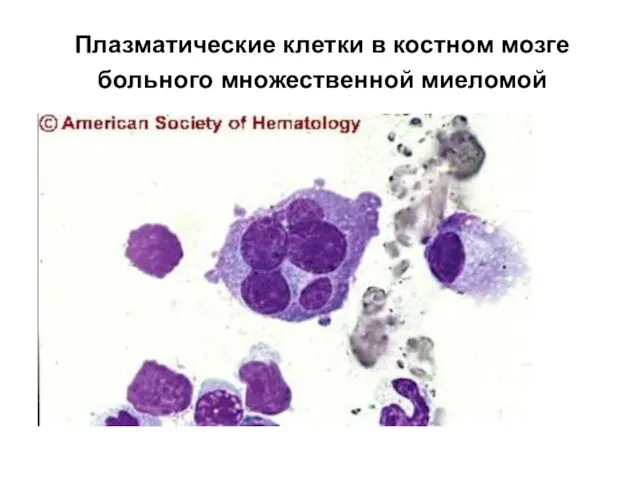
Плазматические клетки в костном мозге больного множественной миеломой
Плазматические клетки в костном мозге больного множественной миеломой

Кортикостероиды
Пульс дексаметазона
VAD
Повторяется каждые 3-4 нед
Декса 40 мг ро день 1-4, 9-12,
Кортикостероиды
Пульс дексаметазона
VAD
Повторяется каждые 3-4 нед
Декса 40 мг ро день 1-4, 9-12,
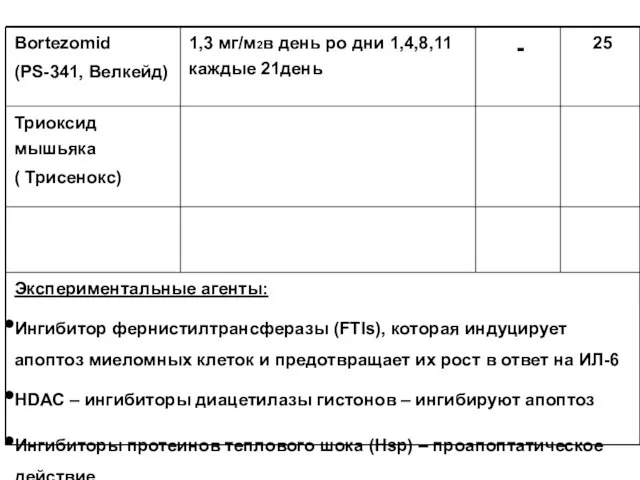
Bortezomid
(PS-341, Велкейд)
1,3 мг/м2в день ро дни 1,4,8,11 каждые 21день
-
25
Триоксид мышьяка
(
Bortezomid
(PS-341, Велкейд)
1,3 мг/м2в день ро дни 1,4,8,11 каждые 21день
-
25
Триоксид мышьяка
(

Лечение осложнений миеломы
Осложнение
Терапевтические подходы
Поражение костей
Бифосфонаты (бонефос, аредиа, зомета)
поощрение двигательной активности для
Лечение осложнений миеломы
Осложнение
Терапевтические подходы
Поражение костей
Бифосфонаты (бонефос, аредиа, зомета)
поощрение двигательной активности для

Анемия
при симптомах анемии эритропоэтин во время химиотерапии
При необходимости - гемотрансфузии
Инфекции
Назначение антибиотиков
Анемия
при симптомах анемии эритропоэтин во время химиотерапии
При необходимости - гемотрансфузии
Инфекции
Назначение антибиотиков

Почечная недостаточ-ность
Коррекция обратимых причин, таких как дегидратация, гиперкальциемия, гиперурикемии
Химиотерапия для быстрого
Почечная недостаточ-ность
Коррекция обратимых причин, таких как дегидратация, гиперкальциемия, гиперурикемии
Химиотерапия для быстрого

Макроглобулинемия Вальденстрема.
Характеризуется наличием моноклонального иммуноглобулина М (PIgM), высокой вязкостью сыворотки
Макроглобулинемия Вальденстрема.
Характеризуется наличием моноклонального иммуноглобулина М (PIgM), высокой вязкостью сыворотки

Клиническая манифестация макроглобулинемии Вальденстрема
Симптомы
Признаки
Слабость, усталость
Симптомы гипервязкости
Ночная потливость
Потеря веса
Периферическая нейропатия
Гепатомегалия - 20%
Спленомегалия
Клиническая манифестация макроглобулинемии Вальденстрема
Симптомы
Признаки
Слабость, усталость
Симптомы гипервязкости
Ночная потливость
Потеря веса
Периферическая нейропатия
Гепатомегалия - 20%
Спленомегалия

Признаки синдрома гипервязкости
Кровотечение из носа и десен, реже – желудочно-кишечные и
Признаки синдрома гипервязкости
Кровотечение из носа и десен, реже – желудочно-кишечные и
 Презентация к 180-летию Альфреда Нобеля
Презентация к 180-летию Альфреда Нобеля Konferentsia_17_02_22 (1)
Konferentsia_17_02_22 (1) Дидактический материал для преодоления нарушения слоговой структуры слова у детей 4-6 лет
Дидактический материал для преодоления нарушения слоговой структуры слова у детей 4-6 лет Европейский удильщик или морской черт
Европейский удильщик или морской черт Воспитание и обучение в Древней Индии
Воспитание и обучение в Древней Индии Потепление в СПБ или Зима без снега
Потепление в СПБ или Зима без снега Irregular verbs. Game
Irregular verbs. Game Основы программирования. Вложенные циклы
Основы программирования. Вложенные циклы Культура России первой половины 19 века
Культура России первой половины 19 века Философия и общественные науки в Новое и Новейшее время
Философия и общественные науки в Новое и Новейшее время Гpузовые стропы общего назначения
Гpузовые стропы общего назначения Происхождение Земли. (Лекция 6)
Происхождение Земли. (Лекция 6) Организация и ведение научно- исследовательской работы среди школьников
Организация и ведение научно- исследовательской работы среди школьников История троллейбуса БТЗ-5276-04
История троллейбуса БТЗ-5276-04 Я - выбираю спорт!
Я - выбираю спорт! Построение сложных запросов. Инсерт
Построение сложных запросов. Инсерт Социальные движения в первой половине XVIII века
Социальные движения в первой половине XVIII века Спасский Староярмарочный Собор. Моя малая Родина. Моё спасение
Спасский Староярмарочный Собор. Моя малая Родина. Моё спасение АНАЛИТИЧЕСКИЙ ОТЧЕТ о результатах итоговых контрольных работ
АНАЛИТИЧЕСКИЙ ОТЧЕТ о результатах итоговых контрольных работ Тема Родительское собрание
Тема Родительское собрание Усилители. Усилительный каскад на БПТ с ОЭ
Усилители. Усилительный каскад на БПТ с ОЭ проект учащегося 3 класса Четверткова Георгия
проект учащегося 3 класса Четверткова Георгия Наши дети – наше будущее
Наши дети – наше будущее Волейбол. Совершенствование нижней прямой подачи
Волейбол. Совершенствование нижней прямой подачи Метод координат
Метод координат Презентация для педагогов
Презентация для педагогов Приобщение детей к татарской и русской культуре в подготовительной группе детского сада
Приобщение детей к татарской и русской культуре в подготовительной группе детского сада Военно-морской флот Российской Федерации
Военно-морской флот Российской Федерации