- Регистрационное досье и документация по GMP - Европейские требования

Содержание
- 2. Регистрация и GMP в ЕС Важнейшие элементы системы обеспечения качества лекарств на национальном уровне
- 3. Регистрация и GMP в ЕС Какой элемент главнее? Традиционно в СССР и в первые годы суверенной
- 4. Регистрация и GMP в ЕС Лаборатории Фармакопеи Инспекции GMP Регистрация Фактически в центре системы процесс регистрации
- 5. Регистрация и GMP в ЕС Позиция международных экспертов – 1985 г. Создать (или укрепить существующий) орган
- 6. Регистрация и GMP в ЕС Качество лекарственного продукта: «трехпалубное» определение Пригодность к применению (по назначению) Соответствие
- 7. Регистрация и GMP в ЕС Формат регистрационного досье - системообразующий элемент Порядок регистрации и, прежде всего,
- 8. Регистрация и GMP в ЕС Лаборатории Фармакопеи Инспекции GMP ОТД CTD-Q Контуры современной системы обеспечения фармацевтического
- 9. Регистрация и GMP в ЕС Регистрационное досье или регистрационные материалы? Досье четко структурировано Разделы и подразделы
- 10. Регистрация и GMP в ЕС Общий технический документ: источники информации «Фармацевтический сектор: Общий технический документ для
- 11. Регистрация и GMP в ЕС Общий технический документ ICH М4 Модуль 1 Региональная административная информация Оглавление
- 12. Регистрация и GMP в ЕС Модуль 1 Административная (региональная) информация • 1.1 Оглавление • 1.2 Форма
- 13. Регистрация и GMP в ЕС Модуль 2 Общий обзор раздела “Качество” (то, что будут читать в
- 14. Регистрация и GMP в ЕС Модуль 3 Качество 3.1 Оглавление Модуля 3 3.2 Регистрационные материалы: S:
- 15. Регистрация и GMP в ЕС 3.2.S.1- Общая информация о субстанции 3.2.S.1.1 Номенклатура: - ИНН, другое непатентованное
- 16. Регистрация и GMP в ЕС Отступление: важнейшие документы ICH Q1 Стабильность Q2(R1) Валидация аналитических методик Q3A(R2)
- 17. Регистрация и GMP в ЕС 3.2.S.1. - Общая информация (для новых субстанций) 3.2.S.1.2 Структура Структурная формула,
- 18. Регистрация и GMP в ЕС 3.2.S.2 - Производство субстанции 3.2.S 2.1 производитель (производители) 3.2.S 2.2 описание
- 19. Регистрация и GMP в ЕС Отступление: active substance master file Дженериковый производитель не располагает данными об
- 20. Регистрация и GMP в ЕС 3.2.S.2 - Производство субстанции - продолжение 3.2.S2.6 разработка производственного процесса: описание
- 21. Регистрация и GMP в ЕС 3.2.S.2 - Производство субстанции - примечание Для субстанций, получаемых биотехнологическим путем
- 22. Регистрация и GMP в ЕС 3.2.S.3 - Характеризация (для новых субстанций) 3.2.S.3.1 выяснение структуры и других
- 23. Регистрация и GMP в ЕС 3.2.S.3.2 - Примеси Органические примеси Неорганические примеси Следы растворителей
- 24. Регистрация и GMP в ЕС 3.2.S.3.2 - Примеси - продолжение Информация о примесях в соответствии с
- 25. Регистрация и GMP в ЕС Примеси - расшифровка Неорганические примеси (как правило, идентифицированные): Реактивы, катализаторы и
- 26. Регистрация и GMP в ЕС 3.2.S.4 - Контроль субстанций (нумерация подразделов опущена) Спецификации Q6A, Q6B аналитические
- 27. Регистрация и GMP в ЕС 3.2.S.5 - Стандартные образцы Информация о стандартных образцах, использованных для анализа
- 28. Регистрация и GMP в ЕС 3.2.S.6 - Упаковочно-укупорочная система Описание упаковочно-укупорочной системы, включая спецификации всех материалов
- 29. Регистрация и GMP в ЕС 3.2.S.7 - Стабильность Обзор и выводы по стабильности Q1A, Q1B, Q5С
- 30. Регистрация и GMP в ЕС 3.2.Р.1 - Описание и состав лекарственного продукта Описание лекарственной формы Q6A,
- 31. Регистрация и GMP в ЕС Описание и состав лекарственного продукта (расшифровка) описание лекформы состав на одну
- 32. Регистрация и GMP в ЕС 3.2.Р.2 - Фармацевтическая разработка Цель ФР - создать качественный продукт и
- 33. Регистрация и GMP в ЕС Фармацевтическая разработка - продолжение Свойства субстанций, влияющие на качество продукта обоснование
- 34. Регистрация и GMP в ЕС 3.2.Р.3 - Производство готового продукта Производитель (производители) название, адрес и сфера
- 35. Регистрация и GMP в ЕС 3.2.Р.3.3 - Описание технологического процесса Технологическая схема, отражающая стадии процесса, с
- 36. Регистрация и GMP в ЕС 3.2.Р.3.4 - Контроль критических этапов и полупродуктов Критические этапы: Испытания и
- 37. Регистрация и GMP в ЕС 3.2.Р.3.5 - Валидация или оценка процессов Описание, документация и результаты валидационных
- 38. Регистрация и GMP в ЕС 3.2.Р.4 - Контроль вспомогательных веществ Спецификации Q6А, Q6B Аналитические методы Q2А,
- 39. Регистрация и GMP в ЕС Р.4 - Контроль вспомогательных веществ продолжение Вспомогатедльные вещества человеческого или животного
- 40. Регистрация и GMP в ЕС 3.2.Р.5 - Контроль лекарственного продукта Спецификация (и) Q3В, Q6А, Q6B Аналитические
- 41. Регистрация и GMP в ЕС 3.2. Р.6 - Стандартные образцы Стандартные образцы или материалы - если
- 42. Регистрация и GMP в ЕС 3.2. Р.7 - Упаковочно- укупорочная система Описание упаковочно-укупорочной системы, включая название
- 43. Регистрация и GMP в ЕС 3.2. Р.8 - Стабильность Р 8.1 Обзор и выводы по стабильности
- 44. Регистрация и GMP в ЕС А - Приложения А 1 Здания и оборудование (для биотехнологических препаратов)
- 46. Скачать презентацию

Регистрация и GMP в ЕС
Важнейшие элементы системы обеспечения качества лекарств
на национальном
Регистрация и GMP в ЕС
Важнейшие элементы системы обеспечения качества лекарств на национальном

Регистрация и GMP в ЕС
Какой элемент главнее?
Традиционно в СССР и в
Регистрация и GMP в ЕС
Какой элемент главнее?
Традиционно в СССР и в
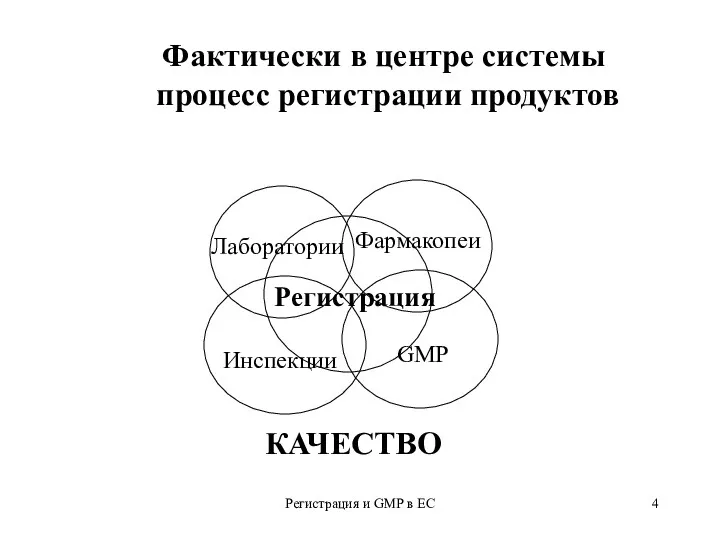
Регистрация и GMP в ЕС
Лаборатории
Фармакопеи
Инспекции
GMP
Регистрация
Фактически в центре системы
Регистрация и GMP в ЕС
Лаборатории
Фармакопеи
Инспекции
GMP
Регистрация
Фактически в центре системы

Регистрация и GMP в ЕС
Позиция международных экспертов – 1985 г.
Создать
Регистрация и GMP в ЕС
Позиция международных экспертов – 1985 г.
Создать

Регистрация и GMP в ЕС
Качество лекарственного продукта:
«трехпалубное» определение
Пригодность к применению
Регистрация и GMP в ЕС
Качество лекарственного продукта:
«трехпалубное» определение
Пригодность к применению

Регистрация и GMP в ЕС
Формат регистрационного досье -
системообразующий элемент
Порядок регистрации и,
Регистрация и GMP в ЕС
Формат регистрационного досье -
системообразующий элемент
Порядок регистрации и,
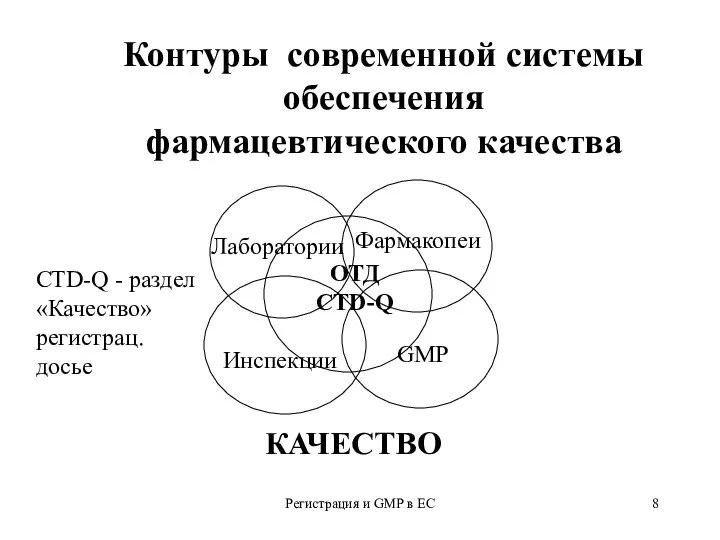
Регистрация и GMP в ЕС
Лаборатории
Фармакопеи
Инспекции
GMP
ОТД
CTD-Q
Контуры современной системы
обеспечения
фармацевтического
Регистрация и GMP в ЕС
Лаборатории
Фармакопеи
Инспекции
GMP
ОТД
CTD-Q
Контуры современной системы
обеспечения
фармацевтического

Регистрация и GMP в ЕС
Регистрационное досье или регистрационные материалы?
Досье четко структурировано
Разделы
Регистрация и GMP в ЕС
Регистрационное досье или регистрационные материалы?
Досье четко структурировано
Разделы

Регистрация и GMP в ЕС
Общий технический документ:
источники информации
«Фармацевтический сектор: Общий
Регистрация и GMP в ЕС
Общий технический документ:
источники информации
«Фармацевтический сектор: Общий
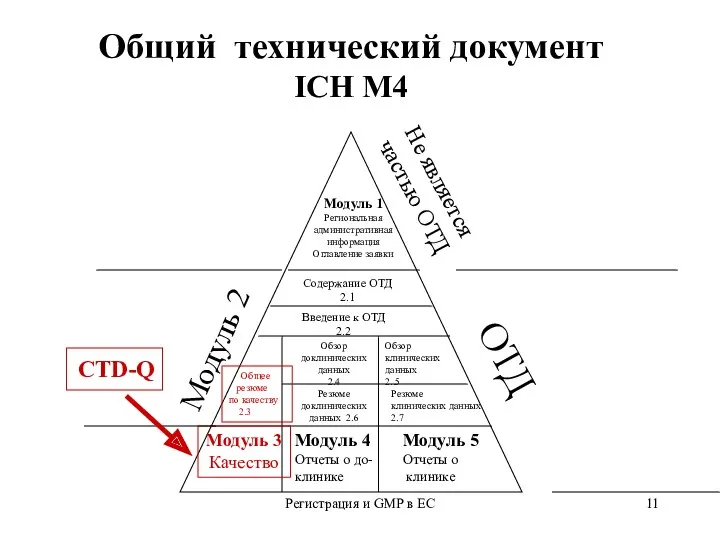
Регистрация и GMP в ЕС
Общий технический документ
ICH М4
Модуль 1
Региональная
административная
информация
Оглавление заявки
Не является
Регистрация и GMP в ЕС
Общий технический документ
ICH М4
Модуль 1
Региональная
административная
информация
Оглавление заявки
Не является

Регистрация и GMP в ЕС
Модуль 1
Административная (региональная) информация
• 1.1 Оглавление
•
Регистрация и GMP в ЕС
Модуль 1
Административная (региональная) информация
• 1.1 Оглавление
•

Регистрация и GMP в ЕС
Модуль 2
Общий обзор раздела “Качество”
(то,
Регистрация и GMP в ЕС
Модуль 2 Общий обзор раздела “Качество” (то,

Регистрация и GMP в ЕС
Модуль 3
Качество
3.1 Оглавление Модуля 3
3.2 Регистрационные материалы:
S:
Регистрация и GMP в ЕС
Модуль 3
Качество
3.1 Оглавление Модуля 3
3.2 Регистрационные материалы:
S:

Регистрация и GMP в ЕС
3.2.S.1- Общая информация о субстанции
3.2.S.1.1 Номенклатура: -
Регистрация и GMP в ЕС
3.2.S.1- Общая информация о субстанции
3.2.S.1.1 Номенклатура: -

Регистрация и GMP в ЕС
Отступление:
важнейшие документы ICH
Q1 Стабильность
Q2(R1) Валидация аналитических методик
Q3A(R2)
Регистрация и GMP в ЕС
Отступление:
важнейшие документы ICH
Q1 Стабильность
Q2(R1) Валидация аналитических методик
Q3A(R2)

Регистрация и GMP в ЕС
3.2.S.1. - Общая информация
(для новых субстанций)
3.2.S.1.2 Структура
Структурная
Регистрация и GMP в ЕС
3.2.S.1. - Общая информация
(для новых субстанций)
3.2.S.1.2 Структура
Структурная

Регистрация и GMP в ЕС
3.2.S.2 - Производство субстанции
3.2.S 2.1 производитель (производители)
3.2.S
Регистрация и GMP в ЕС
3.2.S.2 - Производство субстанции
3.2.S 2.1 производитель (производители)
3.2.S

Регистрация и GMP в ЕС
Отступление:
active substance master file
Дженериковый производитель не
Регистрация и GMP в ЕС
Отступление:
active substance master file
Дженериковый производитель не

Регистрация и GMP в ЕС
3.2.S.2 - Производство субстанции - продолжение
3.2.S2.6 разработка
Регистрация и GMP в ЕС
3.2.S.2 - Производство субстанции - продолжение
3.2.S2.6 разработка

Регистрация и GMP в ЕС
3.2.S.2 - Производство субстанции - примечание
Для субстанций,
Регистрация и GMP в ЕС
3.2.S.2 - Производство субстанции - примечание
Для субстанций,

Регистрация и GMP в ЕС
3.2.S.3 - Характеризация
(для новых субстанций)
3.2.S.3.1 выяснение структуры
Регистрация и GMP в ЕС
3.2.S.3 - Характеризация
(для новых субстанций)
3.2.S.3.1 выяснение структуры

Регистрация и GMP в ЕС
3.2.S.3.2 - Примеси
Органические примеси
Неорганические примеси
Следы
Регистрация и GMP в ЕС
3.2.S.3.2 - Примеси
Органические примеси
Неорганические примеси
Следы

Регистрация и GMP в ЕС
3.2.S.3.2 - Примеси - продолжение
Информация о примесях
Регистрация и GMP в ЕС
3.2.S.3.2 - Примеси - продолжение
Информация о примесях

Регистрация и GMP в ЕС
Примеси - расшифровка
Неорганические примеси (как правило,
Регистрация и GMP в ЕС
Примеси - расшифровка
Неорганические примеси (как правило,

Регистрация и GMP в ЕС
3.2.S.4 - Контроль субстанций
(нумерация подразделов опущена)
Спецификации
Регистрация и GMP в ЕС
3.2.S.4 - Контроль субстанций
(нумерация подразделов опущена)
Спецификации

Регистрация и GMP в ЕС
3.2.S.5 - Стандартные образцы
Информация о стандартных образцах,
использованных
Регистрация и GMP в ЕС
3.2.S.5 - Стандартные образцы
Информация о стандартных образцах,
использованных

Регистрация и GMP в ЕС
3.2.S.6 - Упаковочно-укупорочная система
Описание упаковочно-укупорочной системы,
Регистрация и GMP в ЕС
3.2.S.6 - Упаковочно-укупорочная система
Описание упаковочно-укупорочной системы,

Регистрация и GMP в ЕС
3.2.S.7 - Стабильность
Обзор и выводы по стабильности
Регистрация и GMP в ЕС
3.2.S.7 - Стабильность
Обзор и выводы по стабильности

Регистрация и GMP в ЕС
3.2.Р.1 - Описание и состав лекарственного продукта
Описание
Регистрация и GMP в ЕС
3.2.Р.1 - Описание и состав лекарственного продукта
Описание

Регистрация и GMP в ЕС
Описание и состав лекарственного продукта
(расшифровка)
описание лекформы
состав
Регистрация и GMP в ЕС
Описание и состав лекарственного продукта
(расшифровка)
описание лекформы
состав

Регистрация и GMP в ЕС
3.2.Р.2 - Фармацевтическая
разработка
Цель ФР - создать качественный
Регистрация и GMP в ЕС
3.2.Р.2 - Фармацевтическая
разработка
Цель ФР - создать качественный

Регистрация и GMP в ЕС
Фармацевтическая
разработка - продолжение
Свойства субстанций, влияющие на качество
Регистрация и GMP в ЕС
Фармацевтическая
разработка - продолжение
Свойства субстанций, влияющие на качество

Регистрация и GMP в ЕС
3.2.Р.3 - Производство готового продукта
Производитель (производители) название,
Регистрация и GMP в ЕС
3.2.Р.3 - Производство готового продукта
Производитель (производители) название,

Регистрация и GMP в ЕС
3.2.Р.3.3 - Описание технологического процесса
Технологическая схема, отражающая
Регистрация и GMP в ЕС
3.2.Р.3.3 - Описание технологического процесса
Технологическая схема, отражающая

Регистрация и GMP в ЕС
3.2.Р.3.4 - Контроль критических этапов и полупродуктов
Критические
Регистрация и GMP в ЕС
3.2.Р.3.4 - Контроль критических этапов и полупродуктов
Критические

Регистрация и GMP в ЕС
3.2.Р.3.5 - Валидация или оценка процессов
Описание, документация
Регистрация и GMP в ЕС
3.2.Р.3.5 - Валидация или оценка процессов
Описание, документация

Регистрация и GMP в ЕС
3.2.Р.4 - Контроль вспомогательных веществ
Спецификации Q6А, Q6B
Аналитические
Регистрация и GMP в ЕС
3.2.Р.4 - Контроль вспомогательных веществ
Спецификации Q6А, Q6B
Аналитические

Регистрация и GMP в ЕС
Р.4 - Контроль вспомогательных веществ
продолжение
Вспомогатедльные вещества
Регистрация и GMP в ЕС
Р.4 - Контроль вспомогательных веществ
продолжение
Вспомогатедльные вещества

Регистрация и GMP в ЕС
3.2.Р.5 - Контроль лекарственного продукта
Спецификация (и) Q3В,
Регистрация и GMP в ЕС
3.2.Р.5 - Контроль лекарственного продукта
Спецификация (и) Q3В,

Регистрация и GMP в ЕС
3.2. Р.6 - Стандартные образцы
Стандартные образцы или
Регистрация и GMP в ЕС
3.2. Р.6 - Стандартные образцы
Стандартные образцы или

Регистрация и GMP в ЕС
3.2. Р.7 - Упаковочно-
укупорочная система
Описание упаковочно-укупорочной системы,
Регистрация и GMP в ЕС
3.2. Р.7 - Упаковочно-
укупорочная система
Описание упаковочно-укупорочной системы,

Регистрация и GMP в ЕС
3.2. Р.8 - Стабильность
Р 8.1 Обзор и
Регистрация и GMP в ЕС
3.2. Р.8 - Стабильность
Р 8.1 Обзор и

Регистрация и GMP в ЕС
А - Приложения
А 1 Здания и оборудование
Регистрация и GMP в ЕС
А - Приложения
А 1 Здания и оборудование
 Суб’єкти відповідальності за корупційні правопорушення
Суб’єкти відповідальності за корупційні правопорушення Международный терроризм
Международный терроризм Типология государства. Тема 5
Типология государства. Тема 5 Хозяйственные общества. Общество с ограниченной ответственностью
Хозяйственные общества. Общество с ограниченной ответственностью Инструкция по заполнению акта приема-передачи приза
Инструкция по заполнению акта приема-передачи приза Модели использования культурных ресурсов
Модели использования культурных ресурсов Недействительность сделок. Ничтожные и оспоримые сделки
Недействительность сделок. Ничтожные и оспоримые сделки 20 ноября – Всемирный день ребёнка
20 ноября – Всемирный день ребёнка RIP. Презентація для регіонів
RIP. Презентація для регіонів Методы управления в органах внутренних дел. Тема №4
Методы управления в органах внутренних дел. Тема №4 Правительство РФ
Правительство РФ Передача личных данных
Передача личных данных Международная декларация прав человека
Международная декларация прав человека Основы гражданского права
Основы гражданского права Турнир знатоков права
Турнир знатоков права Принципы гражданского процессуального права
Принципы гражданского процессуального права Загальне вчення про договір. Основи римського права. Лекція 6
Загальне вчення про договір. Основи римського права. Лекція 6 Організація роботи органів та посадових осіб місцевого самоврядування
Організація роботи органів та посадових осіб місцевого самоврядування Защита прав потребителей
Защита прав потребителей Государство и право. Их роль в жизни общества
Государство и право. Их роль в жизни общества Этика речи защитника
Этика речи защитника Семейный кодекс РФ
Семейный кодекс РФ Курсовая работа по теме: Понятие и признаки индивидуального предпринимательства
Курсовая работа по теме: Понятие и признаки индивидуального предпринимательства Понятие преступления.(Лекция№3)
Понятие преступления.(Лекция№3) Договор подряда и его разновидности
Договор подряда и его разновидности Административная ответственность
Административная ответственность Фирменное наименование
Фирменное наименование Коллективный договор
Коллективный договор