- Биоинформатика. Поиск гомологов в базах данных. (Тема 5)

Содержание
- 2. Поиск гомологов в базах даных BLAST FASTA
- 3. При аналізі первинних структур процедура вирівнювання виявляє сходство між послідовностями (sequence similarity), яке може свідчити про
- 4. Гомологичные последовательности – последовательности, имеющие общее происхождение (общего предка). Признаки гомологичности белков сходная 3D-структура в той
- 5. Что изображено? Название последовательности Номер столбца выравнивания Номер последнего в строке остатка ИЗ ЭТОЙ ПОСЛЕДОВАТЕЛЬНОСТИ Консервативный
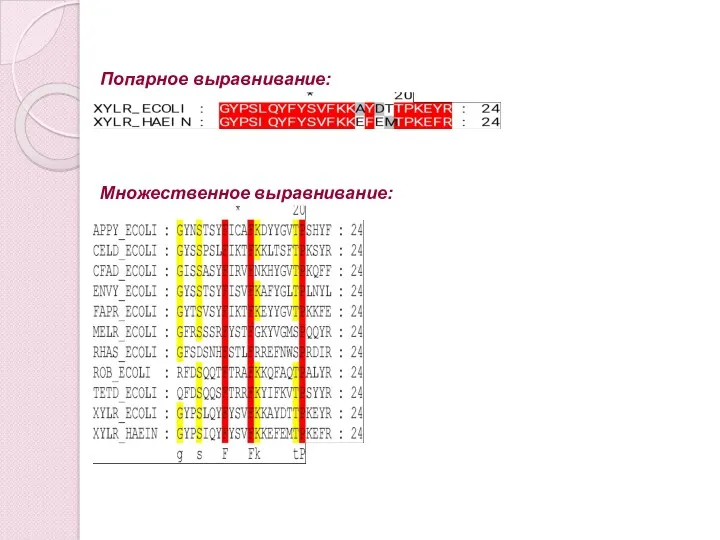
- 6. «Идеальное» выравнивание – запись последовательностей одна под другой так, чтобы гомологичные фрагменты оказались друг под другом.
- 8. Ортологи и паралоги Ортологи – гени з різних організмів, що розійшлися при видоутворенні. Мається на увазі,
- 9. BLAST Что такое выравнивание Выравнивание 2х последовательностей BLAST на NCBI: Что это такое Как выбрать правильную
- 10. Почему нас интересует локальное сходство последовательностей? Мы верим, что: 1. функцию, структуру и многие другие свойства
- 11. Гомологи Ортологи Паралоги Ксенологи ? (W.M.Fitch, Syst.Zool.19,99(1970)
- 12. Схожие 3D структуры Вставка в «синей» последовательности
- 13. Матрицы замен Матрица 20*20 на пересечении 2х aa их уровень сходства (?): Похожесть по свойствам (объем,
- 14. Делеции/инсерции Общий штраф Значительно чаще 1 длинная делеция, чем много коротких => штраф за внесение делеции
- 15. Типы выравнивания Локальное – поиск фрагментов наиболее похожих друг на друга домовой домовой домовой скупидом водомерка
- 16. Критерии качества выравнивания Количество идентичных (похожих) аминокислот/нуклеотидов Для белков – более 25% id при длине >
- 17. Поиск гомологов в базах даных FASTA (Pearson and Lipman, 1988) BLAST (Altschul et al., 1990)
- 18. FASTA 1.A lookup table is generated consisting of short stretches of amino acids or nucleotides from
- 19. FASTA http://www.ebi.ac.uk/Tools/sss/fasta/
- 20. BLAST – Basic Local Alignment and Search Tool Локальное выравнивание Главная задача – поиск похожих последовательностей
- 21. Родной BLAST – NCBI (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi)
- 22. Basic Local Alignment Search Tool Также, как FASTA, требует параметр k (длина слова). Белки k= 3
- 23. 1. Поиск идентичных\похожих участков 2. Попытка «удлинить» эти участки насколько возможно (т.е. пока score растёт) В
- 24. Попытка соединить соседние HSPs путем выравнивания последовательностей между ними: THEFIRSTLINIHAVEADREA____M_ESIRPATRICKREAD INVIEIAMDEADMEATTNAMHEW___ASNINETEEN Алгоритм BLAST (шаг 2)
- 25. Blast Blast – это семейство программ: BlastN, BlastP, BlastX, tBlastN BlastN - ДНК vs ДНК BlastP
- 26. Blast
- 27. Одною з розповсюджених прикладних задач є пошук гомологів відомих білків у повністю розшифрованих геномах. пряме співставлення
- 28. Стратегія 1 Ми перетворюємо цільову амінокислотну послідовність в набір нуклеотидних послідовностей, згідно стандартного генетичного коду. На
- 29. Стратегія 2 Ми перетворюємо (транслюємо) вміст нуклеотидної бази даних в амінокислотні послідовності. На виході отримуємо 6
- 30. Поиск гомологов По ДНК или по белку? Какой поиск предпочтительней?
- 31. ДНК или белок? Какая последовательность более постоянна в эволюционном плане? UCAUAC Or Serine -Tyrosine
- 32. Генетический код избыточен – почти все аминокислоты кодируются более, чем 1 кодоном (тройка нуклеотидов) Последовательность ДНК
- 33. Нуклеотиды – 4-х буквенный алфавит. Аминокислоты – 20-и буквенный алфавит Две случайные последовательности ДНК будут идентичны
- 34. Матрицы для сравнения белков более чувствительны, чем матрицы для ДНК. Базы данных ДНК намного больше белковых
- 35. Использование белковых последовательностей более предпочтительно при поиске гомологов Поиск гомологов
- 36. Специализированные инструменты ДНК: megaBLAST – другой алгоритм для сравнения ДНК. Оптимизирован для длинных похожих последовательностей. Оптимален
- 37. Специализированные инструменты ДНК: megaBLAST – другой алгоритм для сравнения ДНК. Оптимизирован для длинных похожих последовательностей. Оптимален
- 38. Какую программу выбрать? BLAST
- 39. Стандартный input
- 40. Промежуточная страница - СD
- 41. Output - I
- 42. Output - II
- 43. Output - III
- 44. Output IV
- 45. E-value, bit score E-value (математическое ожидание, the expectation value) – оценка числа раз наблюдать хит такого
- 47. E-value, bit score Bit Score – мера статистической значимости (вес – сумма стоимостей всех точечных замен)
- 48. Параметры выравнивания Матрица:BLOSUM для локального выравнивания обычно лучше, чем PAM Чем выше номер BLOSUM – тем
- 49. Сообщение о параметрах В конце файла текстовая информация об использованный параметрах: Использованная матрица замен Штрафы за
- 50. Выбор параметров Меняйте параметры только, если по умолчанию не работает (параметры по умолчанию подобраны хорошо для
- 51. Какие параметры менять? Фильтрация Low-complexity region – другой aa-состав Фильтрация: если Ваш белок содержит большой регион
- 52. Параметры output-формата Количество хитов Выбор базы данных (организм) Выбор порога - Expect (если хитов мало, то
- 53. PSI - BLAST Алгоритм: Несколько раундов поиска Первый раунд – просто blastp (BLOSUM62) Построение PSSM на
- 54. PSSM Portion of a PSSM from a PSI-BLAST search using RBP4 (NP_006735) as a query. The
- 55. PHI - BLAST Query – белок + паттерн, которому этот белок удовлетворяет Пример: >P28332|ADH6_HUMAN Alcohol dehydrogenase
- 56. НММ-профиль Hidden Markov models describe alignments based on the probability of amino acids occurring in an
- 57. Пример простого мотива
- 58. Другие программы поиска по БД: FASTA (www.ebi.ac.uk/fasta33/) Ssearch (алгоритм Smith-Waterman) (www.ch.embnet.org) BLAT (genome.ucsc.edu)
- 60. Скачать презентацию

Поиск гомологов в базах даных
BLAST
FASTA
Поиск гомологов в базах даных
BLAST
FASTA

При аналізі первинних структур процедура вирівнювання виявляє сходство між послідовностями (sequence
При аналізі первинних структур процедура вирівнювання виявляє сходство між послідовностями (sequence

Гомологичные последовательности – последовательности, имеющие общее происхождение (общего предка).
Признаки гомологичности
Гомологичные последовательности – последовательности, имеющие общее происхождение (общего предка).
Признаки гомологичности
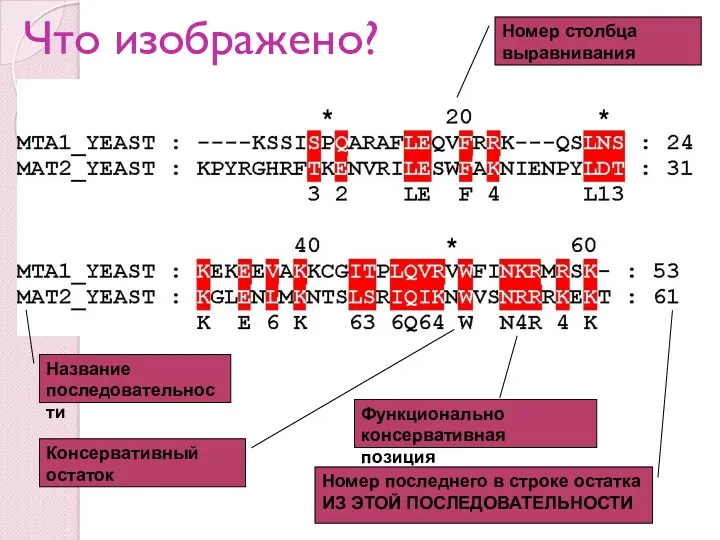
Что изображено?
Название последовательности
Номер столбца выравнивания
Номер последнего в строке остатка ИЗ ЭТОЙ
Что изображено?
Название последовательности
Номер столбца выравнивания
Номер последнего в строке остатка ИЗ ЭТОЙ

«Идеальное» выравнивание – запись последовательностей одна под другой так, чтобы гомологичные
«Идеальное» выравнивание – запись последовательностей одна под другой так, чтобы гомологичные

Ортологи и паралоги
Ортологи – гени з різних організмів, що розійшлися при
Ортологи и паралоги
Ортологи – гени з різних організмів, що розійшлися при

BLAST
Что такое выравнивание
Выравнивание 2х последовательностей
BLAST на NCBI:
Что это такое
Как выбрать
BLAST
Что такое выравнивание
Выравнивание 2х последовательностей
BLAST на NCBI:
Что это такое
Как выбрать

Почему нас интересует локальное сходство последовательностей?
Мы верим, что:
1. функцию, структуру и
Почему нас интересует локальное сходство последовательностей?
Мы верим, что:
1. функцию, структуру и
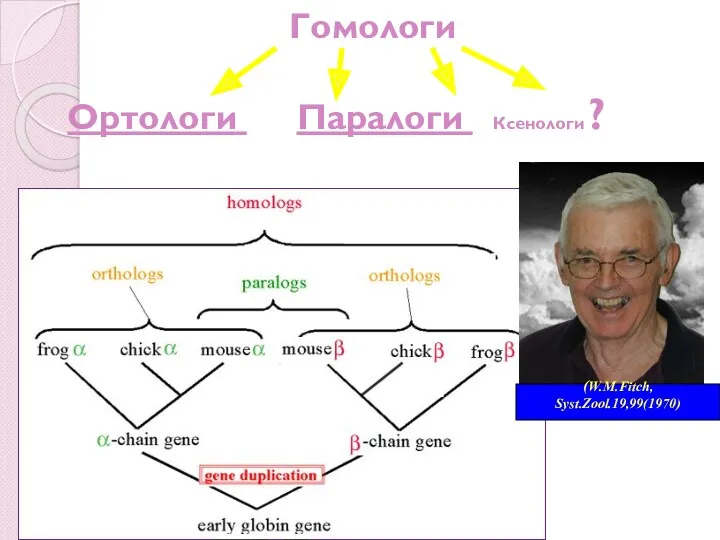
Гомологи
Ортологи Паралоги Ксенологи ?
(W.M.Fitch, Syst.Zool.19,99(1970)
Гомологи
Ортологи Паралоги Ксенологи ?
(W.M.Fitch, Syst.Zool.19,99(1970)
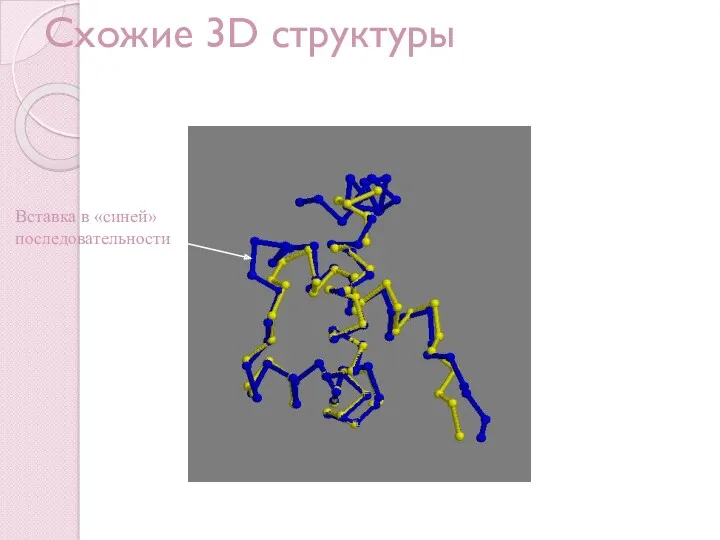
Схожие 3D структуры
Вставка в «синей» последовательности
Схожие 3D структуры
Вставка в «синей» последовательности

Матрицы замен
Матрица 20*20 на пересечении 2х aa их уровень сходства (?):
Похожесть
Матрицы замен
Матрица 20*20 на пересечении 2х aa их уровень сходства (?):
Похожесть

Делеции/инсерции
Общий штраф
Значительно чаще 1 длинная делеция, чем много коротких => штраф
Делеции/инсерции
Общий штраф
Значительно чаще 1 длинная делеция, чем много коротких => штраф
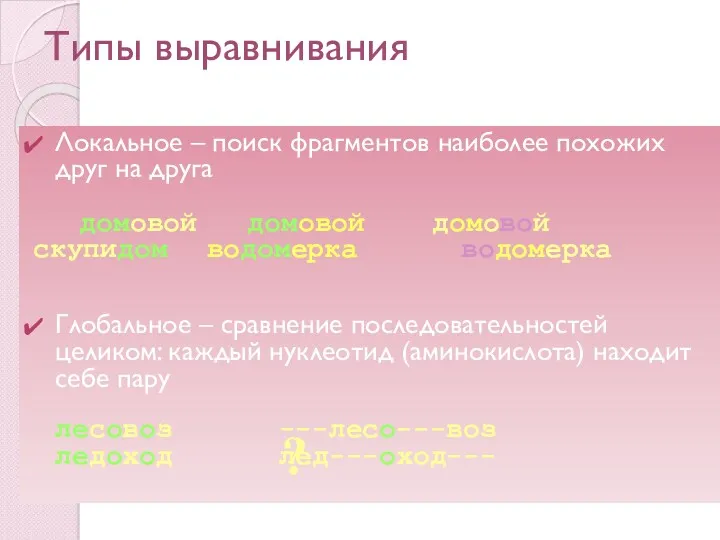
Типы выравнивания
Локальное – поиск фрагментов наиболее похожих друг на друга
домовой домовой
Типы выравнивания
Локальное – поиск фрагментов наиболее похожих друг на друга
домовой домовой

Критерии качества выравнивания
Количество идентичных (похожих) аминокислот/нуклеотидов
Для белков – более 25%
Критерии качества выравнивания
Количество идентичных (похожих) аминокислот/нуклеотидов
Для белков – более 25%

Поиск гомологов в базах даных
FASTA (Pearson and Lipman, 1988)
BLAST (Altschul et
Поиск гомологов в базах даных
FASTA (Pearson and Lipman, 1988)
BLAST (Altschul et

FASTA
1.A lookup table is generated consisting of short stretches of amino
FASTA
1.A lookup table is generated consisting of short stretches of amino

FASTA
http://www.ebi.ac.uk/Tools/sss/fasta/
FASTA
http://www.ebi.ac.uk/Tools/sss/fasta/

BLAST – Basic Local Alignment and Search Tool
Локальное выравнивание
Главная задача –
BLAST – Basic Local Alignment and Search Tool
Локальное выравнивание
Главная задача –
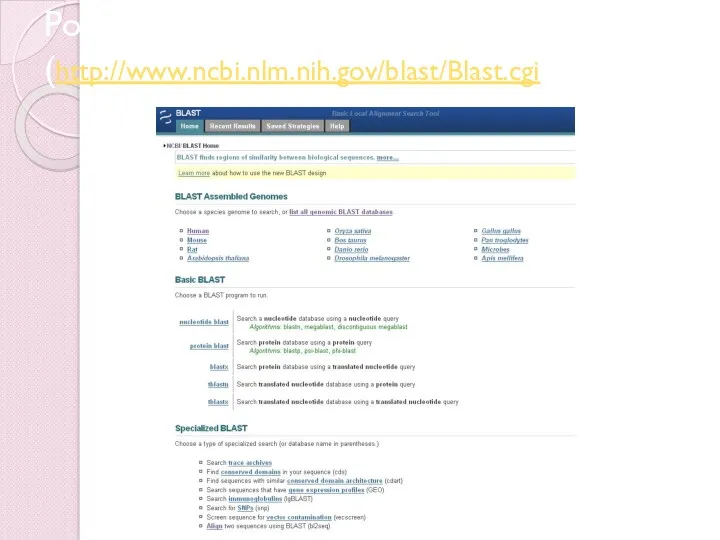
Родной BLAST – NCBI (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi)
Родной BLAST – NCBI (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi)

Basic Local Alignment Search Tool
Также, как FASTA, требует параметр k (длина
Basic Local Alignment Search Tool
Также, как FASTA, требует параметр k (длина

1. Поиск идентичных\похожих участков
2. Попытка «удлинить» эти участки насколько возможно (т.е.
1. Поиск идентичных\похожих участков
2. Попытка «удлинить» эти участки насколько возможно (т.е.

Попытка соединить соседние HSPs путем выравнивания последовательностей между ними:
THEFIRSTLINIHAVEADREA____M_ESIRPATRICKREAD
INVIEIAMDEADMEATTNAMHEW___ASNINETEEN
Алгоритм BLAST
Попытка соединить соседние HSPs путем выравнивания последовательностей между ними:
THEFIRSTLINIHAVEADREA____M_ESIRPATRICKREAD
INVIEIAMDEADMEATTNAMHEW___ASNINETEEN
Алгоритм BLAST

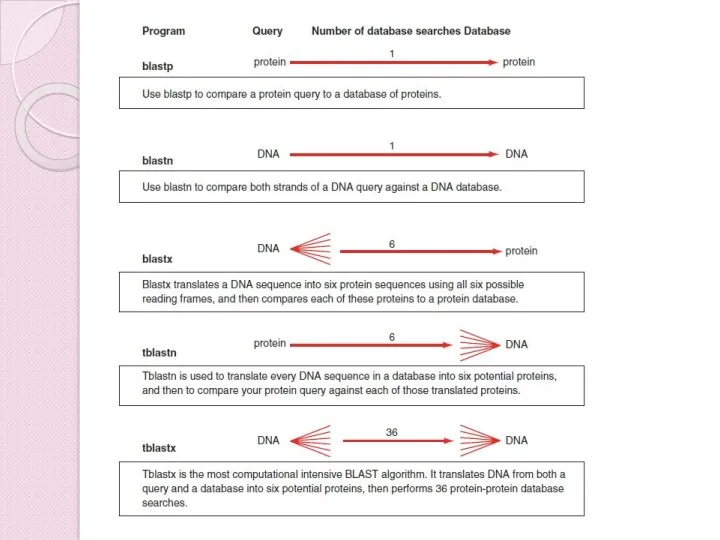
Blast
Blast – это семейство программ: BlastN, BlastP, BlastX, tBlastN
BlastN - ДНК
Blast
Blast – это семейство программ: BlastN, BlastP, BlastX, tBlastN
BlastN - ДНК
Blast
Blast

Одною з розповсюджених прикладних задач є пошук гомологів відомих білків у
Одною з розповсюджених прикладних задач є пошук гомологів відомих білків у

Стратегія 1
Ми перетворюємо цільову амінокислотну послідовність в набір нуклеотидних послідовностей, згідно
Стратегія 1
Ми перетворюємо цільову амінокислотну послідовність в набір нуклеотидних послідовностей, згідно

Стратегія 2
Ми перетворюємо (транслюємо) вміст нуклеотидної бази даних в амінокислотні
Стратегія 2
Ми перетворюємо (транслюємо) вміст нуклеотидної бази даних в амінокислотні

Поиск гомологов
По ДНК или по белку?
Какой поиск предпочтительней?
Поиск гомологов
По ДНК или по белку?
Какой поиск предпочтительней?
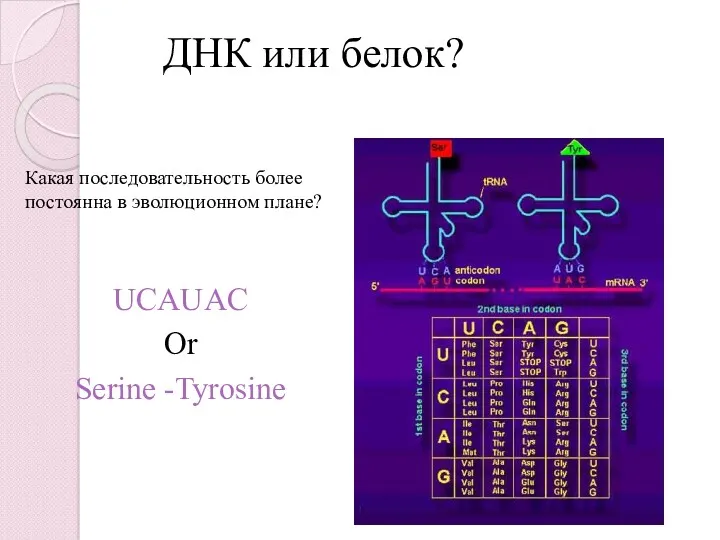
ДНК или белок?
Какая последовательность более постоянна в эволюционном плане?
UCAUAC
Or
Serine -Tyrosine
ДНК или белок?
Какая последовательность более постоянна в эволюционном плане?
UCAUAC
Or
Serine -Tyrosine

Генетический код избыточен – почти все аминокислоты кодируются более, чем 1
Генетический код избыточен – почти все аминокислоты кодируются более, чем 1

Нуклеотиды – 4-х буквенный алфавит.
Аминокислоты – 20-и буквенный алфавит
Две случайные последовательности
Нуклеотиды – 4-х буквенный алфавит.
Аминокислоты – 20-и буквенный алфавит
Две случайные последовательности

Матрицы для сравнения белков более чувствительны, чем матрицы для ДНК.
Базы
Матрицы для сравнения белков более чувствительны, чем матрицы для ДНК.
Базы

Использование белковых последовательностей более предпочтительно при поиске гомологов
Поиск гомологов
Использование белковых последовательностей более предпочтительно при поиске гомологов
Поиск гомологов

Специализированные инструменты
ДНК:
megaBLAST – другой алгоритм для сравнения ДНК. Оптимизирован для
Специализированные инструменты
ДНК:
megaBLAST – другой алгоритм для сравнения ДНК. Оптимизирован для

Специализированные инструменты
ДНК:
megaBLAST – другой алгоритм для сравнения ДНК. Оптимизирован для
Специализированные инструменты
ДНК:
megaBLAST – другой алгоритм для сравнения ДНК. Оптимизирован для

Какую программу выбрать?
BLAST
Какую программу выбрать?
BLAST
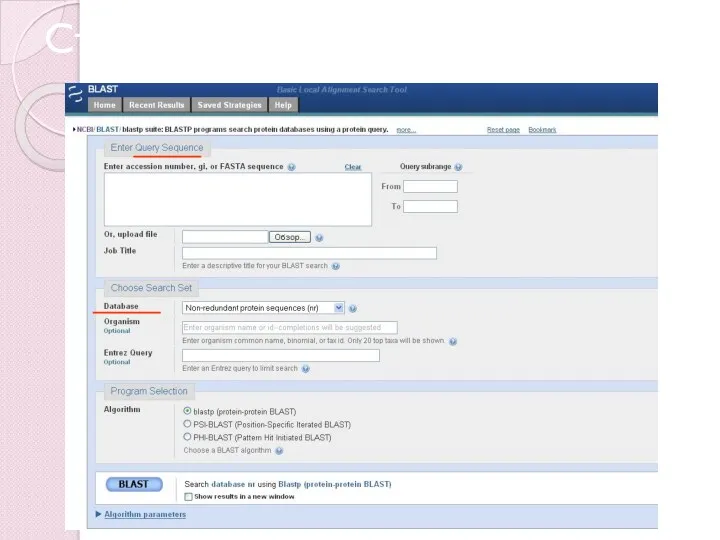
Стандартный input
Стандартный input
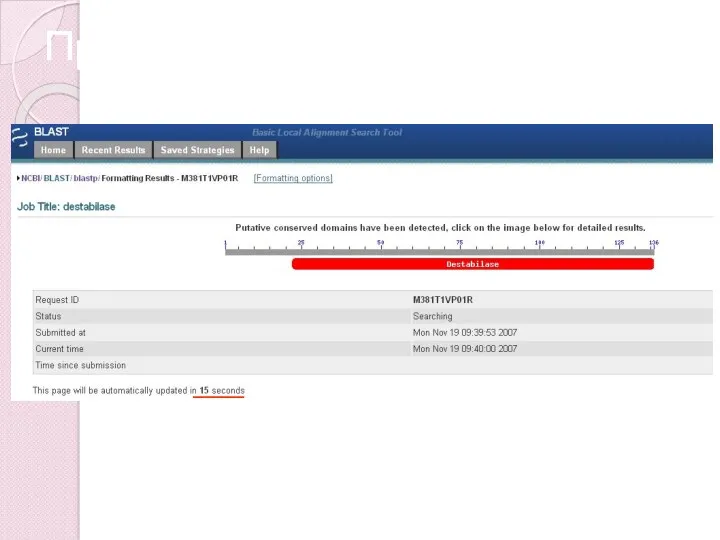
Промежуточная страница - СD
Промежуточная страница - СD
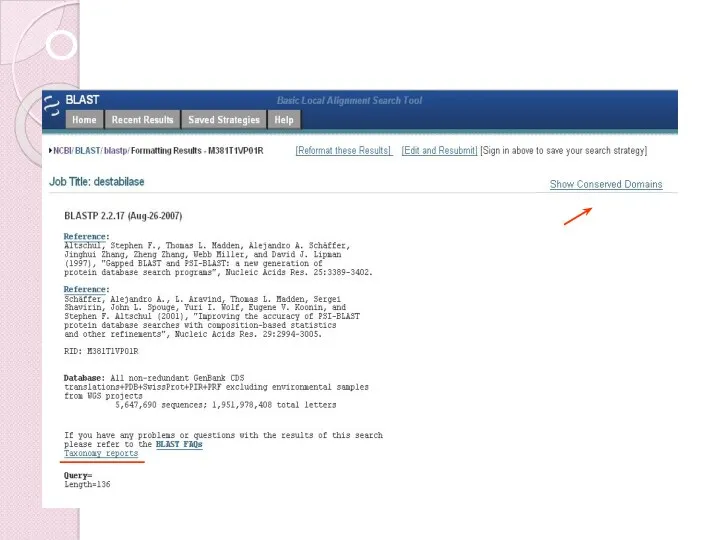
Output - I
Output - I
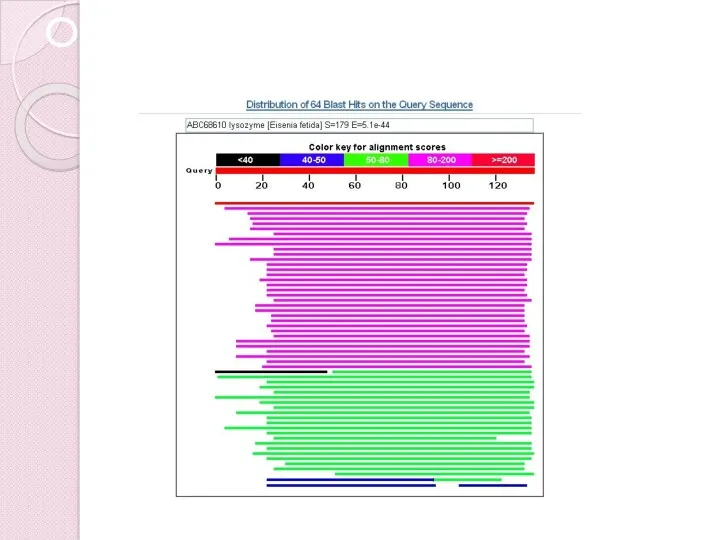
Output - II
Output - II

Output - III
Output - III

Output IV
Output IV

E-value, bit score
E-value (математическое ожидание, the expectation value) – оценка числа
E-value, bit score
E-value (математическое ожидание, the expectation value) – оценка числа


E-value, bit score
Bit Score – мера статистической значимости (вес – сумма
E-value, bit score
Bit Score – мера статистической значимости (вес – сумма

Параметры выравнивания
Матрица:BLOSUM для локального выравнивания обычно лучше, чем PAM
Чем выше номер
Параметры выравнивания
Матрица:BLOSUM для локального выравнивания обычно лучше, чем PAM
Чем выше номер

Сообщение о параметрах
В конце файла текстовая информация об использованный параметрах:
Использованная матрица
Сообщение о параметрах
В конце файла текстовая информация об использованный параметрах:
Использованная матрица

Выбор параметров
Меняйте параметры только, если по умолчанию не работает (параметры по
Выбор параметров
Меняйте параметры только, если по умолчанию не работает (параметры по

Какие параметры менять? Фильтрация
Low-complexity region – другой aa-состав
Фильтрация: если Ваш белок
Какие параметры менять? Фильтрация
Low-complexity region – другой aa-состав
Фильтрация: если Ваш белок

Параметры output-формата
Количество хитов
Выбор базы данных (организм)
Выбор порога - Expect (если хитов
Параметры output-формата
Количество хитов
Выбор базы данных (организм)
Выбор порога - Expect (если хитов

PSI - BLAST
Алгоритм:
Несколько раундов поиска
Первый раунд – просто blastp (BLOSUM62)
Построение
PSI - BLAST
Алгоритм:
Несколько раундов поиска
Первый раунд – просто blastp (BLOSUM62)
Построение

PSSM
Portion of a PSSM from a PSI-BLAST search using RBP4 (NP_006735)
PSSM
Portion of a PSSM from a PSI-BLAST search using RBP4 (NP_006735)

PHI - BLAST
Query – белок + паттерн, которому этот белок удовлетворяет
Пример:
>P28332|ADH6_HUMAN
PHI - BLAST
Query – белок + паттерн, которому этот белок удовлетворяет
Пример:
>P28332|ADH6_HUMAN

НММ-профиль
Hidden Markov models describe alignments based on the probability of amino
НММ-профиль
Hidden Markov models describe alignments based on the probability of amino

Пример простого мотива
Пример простого мотива

Другие программы поиска по БД:
FASTA (www.ebi.ac.uk/fasta33/)
Ssearch (алгоритм Smith-Waterman) (www.ch.embnet.org)
BLAT (genome.ucsc.edu)
Другие программы поиска по БД:
FASTA (www.ebi.ac.uk/fasta33/)
Ssearch (алгоритм Smith-Waterman) (www.ch.embnet.org)
BLAT (genome.ucsc.edu)
 Тварини та їхні особливості. Жираф - найвища тварина на Землі
Тварини та їхні особливості. Жираф - найвища тварина на Землі Презентация к уроку биологии 7 класс
Презентация к уроку биологии 7 класс Обмен веществ и энергии. Терморегуляция. Лекция
Обмен веществ и энергии. Терморегуляция. Лекция Особенности физиологии человека в условиях высокого и низкого давления
Особенности физиологии человека в условиях высокого и низкого давления Мимические мышцы лица
Мимические мышцы лица Положение человека в системе животного мира. Стадии антропогенеза
Положение человека в системе животного мира. Стадии антропогенеза Cerphidae. Ден
Cerphidae. Ден Зоология позвоночных. Строение основных типов беспозвоночных
Зоология позвоночных. Строение основных типов беспозвоночных Красноухая черепаха
Красноухая черепаха Основи механізованої технології виробництва культур. Лекция №7
Основи механізованої технології виробництва культур. Лекция №7 Органы выделения
Органы выделения Природные зоны
Природные зоны Мезозойская эра
Мезозойская эра Физиология гладких мышц. (Лекция 7)
Физиология гладких мышц. (Лекция 7) Презентация Открытия в области биотехнологии
Презентация Открытия в области биотехнологии Уразовский Заказник
Уразовский Заказник Презентация по теме Основные типы экологических взаимодействий
Презентация по теме Основные типы экологических взаимодействий Презентация Загрязнение воздуха
Презентация Загрязнение воздуха Факторы влияющие на качество молока. Лекция 4
Факторы влияющие на качество молока. Лекция 4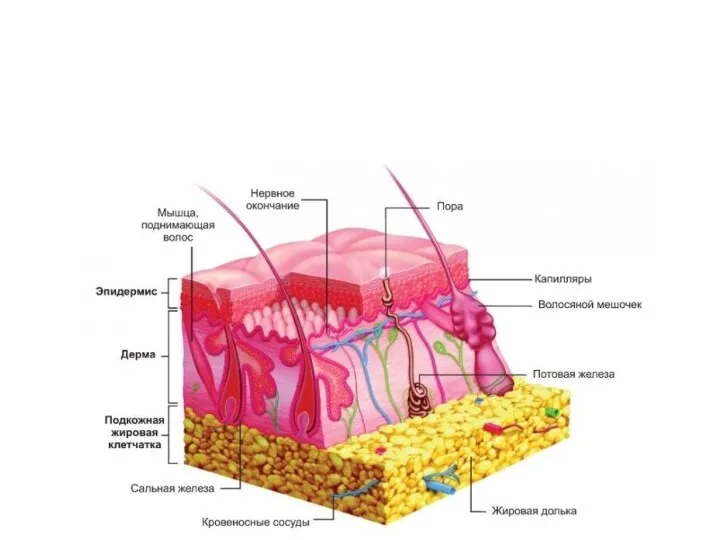 Строение и функции кожи. Наружный слой - эпидермис
Строение и функции кожи. Наружный слой - эпидермис Царство прокариоты, подцарство бактерии
Царство прокариоты, подцарство бактерии Общее представление о биотестировании и биоиндикации
Общее представление о биотестировании и биоиндикации Кольчатые черви
Кольчатые черви Ящерицы
Ящерицы Квітка - вкорочений видозмінений пагін - орган насіннєвого розмноження
Квітка - вкорочений видозмінений пагін - орган насіннєвого розмноження Белки
Белки Внешнее строение рыб. Надкласс рыбы
Внешнее строение рыб. Надкласс рыбы Происхождение современного человека. (Тема 3)
Происхождение современного человека. (Тема 3)