- Болезни с нетрадиционным наследованием

Содержание
- 2. В последние годы стало очевидным, что далеко не все случаи наследственной патологии у человека можно рассматривать
- 3. 1. Геномный импринтинг (ГИ) (импринтинг от англ. imprinting — запечатление). ГИ- эпигенетический процесс, который приводит к
- 4. Явление геномного импринтинга связывают c метилированием цитозиновых оснований ДНК, (выключающим транскрипцию гена) во время образования мужских
- 6. Импринтированные участки в хромосомах определенного родительского происхождения (отцовских иди материнских) избирательно репрессируются у потомка. В связи
- 7. Болезни импринтинга 1. Болезни генного импринтинга 2.Болезни хромосомного импринтинга 3. Болезни геномного импринтинга 4. Болезни ошибок
- 8. 1. Болезни генного импринтинга При болезнях генного импринтинга наблюдается моноаллельная экспрессия в локусах хромосом одного из
- 9. Болезни генного импринтинга К таким заболеваниям относятся: • болезнь Гиршпрунга, обусловленная мутацией в гене RET (10q11.2);
- 10. 2. Болезни хромосомного импринтинга Для болезней хромосомного импринтинга характерна однородительская дисомия (ОРД) - наличие двух копий
- 11. Источники появления однородительской дисомии: 1. Комплиментация гамет – дополнение нуллисомной по определенной хромосоме гаметы, дисомной по
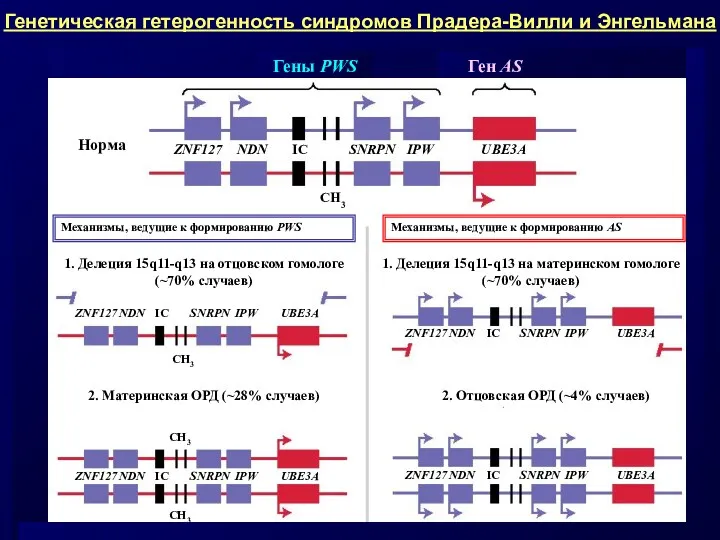
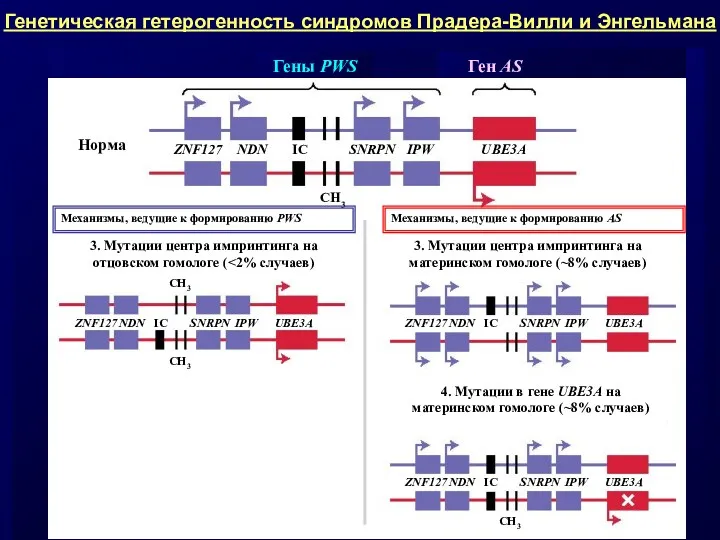
- 12. Примерами таких заболеваний служат: • материнская и отцовская ОРД по хромосоме 15 ведет к синдрому Прадера-Вилли
- 13. Сравнительная характеристика синдромов Прадера-Вилли и Энгельмана
- 18. Синдром Видемана-Беквитта Синдром Беквитта-Видемана (СБВ) относится к распространенным наследственным заболеваниям с частотой в популяции 1:10- 12
- 19. Основные клинические признаки СВБ: гигантизм (масса тела при рождении свыше 3900 г), черепно-лицевой дисморфизм (долихоцефалия, гипоплазия
- 20. Генетика СВБ Критической областью для СБВ является терминальная часть короткого плеча хромосомы 11 (11р15.5). В этой
- 21. Генетика СВБ- Преимущественная потеря материнских аллелей в опухолях, связанных с СВБ или отцовская ОРД по 11p15.5.
- 23. 3. Болезни геномного импринтинга
- 25. 4. Болезни ошибок импринтинга Болезни ошибок импринтинга - результат микроделеций в регуляторных областях импринтированных генов, или
- 27. 2. Митохондриальные болезни Митохондриальный геном человека (мтДНК) состоит из кольцевой ДНК размером 16600 нуклеотидов, которая кодирует
- 28. Митохондриальные заболевания передаются только по женской линии, к детям обоих полов, так как сперматозоиды переносят половину
- 29. Можно выделить 2 группы митохондриальных заболеваний: Ярко выраженные наследственные синдромы, обусловленные мутациями генов, ответственных за митохондриальные
- 30. Синдром MELAS (прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой — диабетом,
- 31. В каждом конкретном случае набор симптомов и их тяжесть может сильно отличаться, поскольку синдром связан с
- 32. НАСЛЕДСТВЕННАЯ ОПТИЧЕСКАЯ НЕЙРОПАТИЯ ЛЕБЕРА Пока описано 19 точковых мутаций в мтДНК.Эти мутации затрагивают компоненты комплексов I
- 33. Пигментный ретинит Задержка развития, умственная отсталость, сенсорная нейропатия, атаксия, нейрогенная мышечная слабость. Развивается в результате точковой
- 34. 3. Болезни экспансии тринуклеотидных повторов (ЭТП) Открытие феномена ЭТП было сделано при изучении синдрома Мартина-Белла (синдром
- 35. Этот синдром характеризуется delXq27.3, умственной отсталостью, аутизмом, оттопыренными ушами, удлиненным лицом, макроорхидизмом.
- 36. Необычность наследования данного синдрома заключается в том, что не все, а только 80% мужчин- носителей данного
- 37. В основе клинических проявления синдрома Мартина-Белла лежит многократное увеличение в первом экзоне гена FMR1 простого тринуклеотидного
- 39. Болезнь Гентингтона - генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 35-50 лет и
- 40. Ген гентингтин (HTT) кодирует белок гентингтин, расположен на коротком плече 4-й хромосомы (4p16.3). Этот ген состоит
- 41. У здоровых людей - от 8 до 25 повторов CAG. Если их становится больше 36, то
- 42. Болезнь Гентингтона поражает специфические области мозга. Одними из первых бывают выразительные движения в виде гримас с
- 43. С момента появления первых симптомов продолжительность жизни составляет около 15–20 лет. Смерть обычно происходит не из-за
- 45. Скачать презентацию

В последние годы стало очевидным, что далеко не все случаи наследственной
В последние годы стало очевидным, что далеко не все случаи наследственной
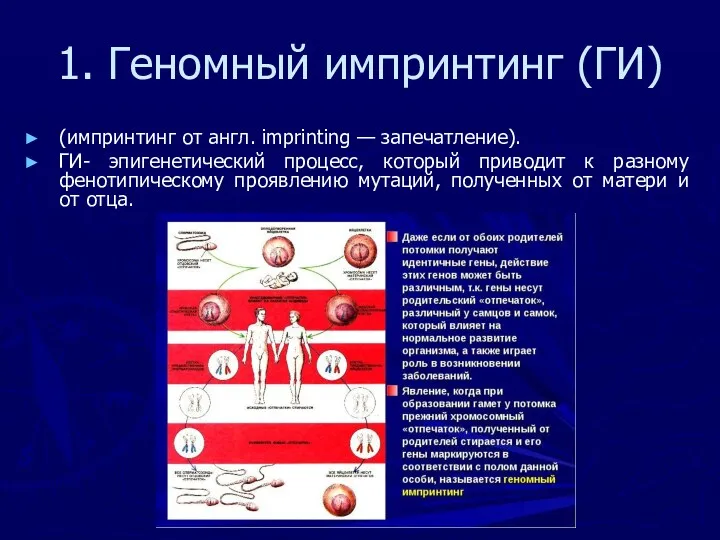
1. Геномный импринтинг (ГИ)
(импринтинг от англ. imprinting — запечатление).
ГИ- эпигенетический процесс,
1. Геномный импринтинг (ГИ)
(импринтинг от англ. imprinting — запечатление).
ГИ- эпигенетический процесс,

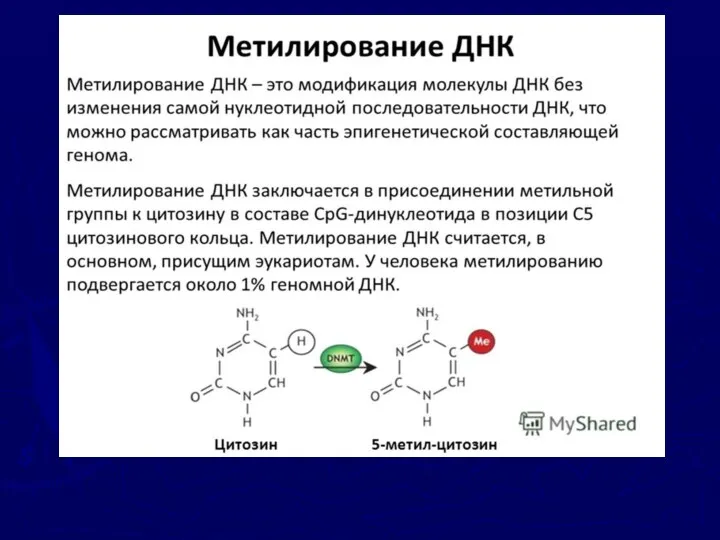
Явление геномного импринтинга связывают c метилированием цитозиновых оснований ДНК, (выключающим транскрипцию
Явление геномного импринтинга связывают c метилированием цитозиновых оснований ДНК, (выключающим транскрипцию

Импринтированные участки в хромосомах определенного родительского происхождения (отцовских иди материнских) избирательно
Импринтированные участки в хромосомах определенного родительского происхождения (отцовских иди материнских) избирательно

Болезни импринтинга
1. Болезни генного импринтинга
2.Болезни хромосомного импринтинга
3. Болезни геномного импринтинга
4. Болезни
Болезни импринтинга
1. Болезни генного импринтинга
2.Болезни хромосомного импринтинга
3. Болезни геномного импринтинга
4. Болезни

1. Болезни генного импринтинга
При болезнях генного импринтинга наблюдается моноаллельная экспрессия в
1. Болезни генного импринтинга
При болезнях генного импринтинга наблюдается моноаллельная экспрессия в

Болезни генного импринтинга
К таким заболеваниям относятся:
• болезнь Гиршпрунга, обусловленная мутацией в
Болезни генного импринтинга
К таким заболеваниям относятся:
• болезнь Гиршпрунга, обусловленная мутацией в

2. Болезни хромосомного импринтинга
Для болезней хромосомного импринтинга характерна однородительская дисомия (ОРД)
2. Болезни хромосомного импринтинга
Для болезней хромосомного импринтинга характерна однородительская дисомия (ОРД)

Источники появления однородительской дисомии:
1. Комплиментация гамет – дополнение нуллисомной по определенной
Источники появления однородительской дисомии:
1. Комплиментация гамет – дополнение нуллисомной по определенной

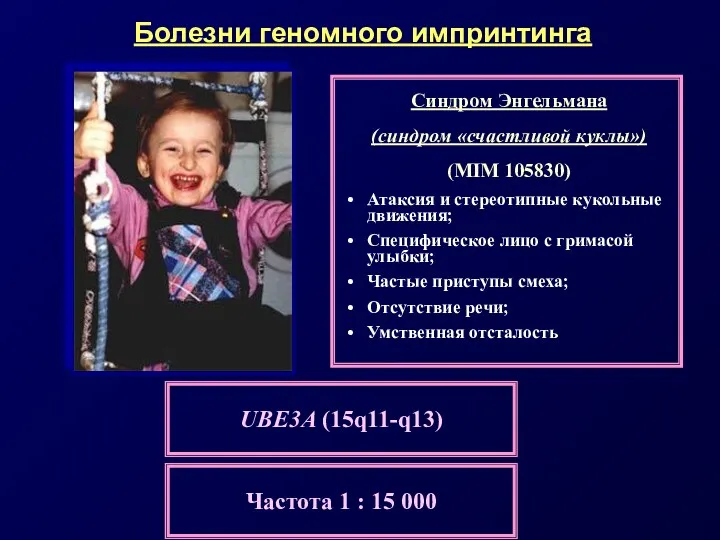
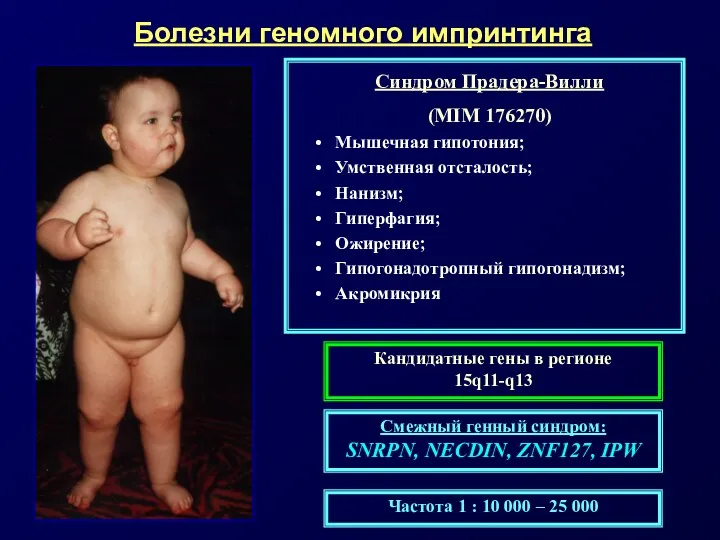
Примерами таких заболеваний служат:
• материнская и отцовская ОРД по хромосоме 15
Примерами таких заболеваний служат:
• материнская и отцовская ОРД по хромосоме 15

Сравнительная характеристика синдромов Прадера-Вилли и Энгельмана
Сравнительная характеристика синдромов Прадера-Вилли и Энгельмана
Синдром Видемана-Беквитта
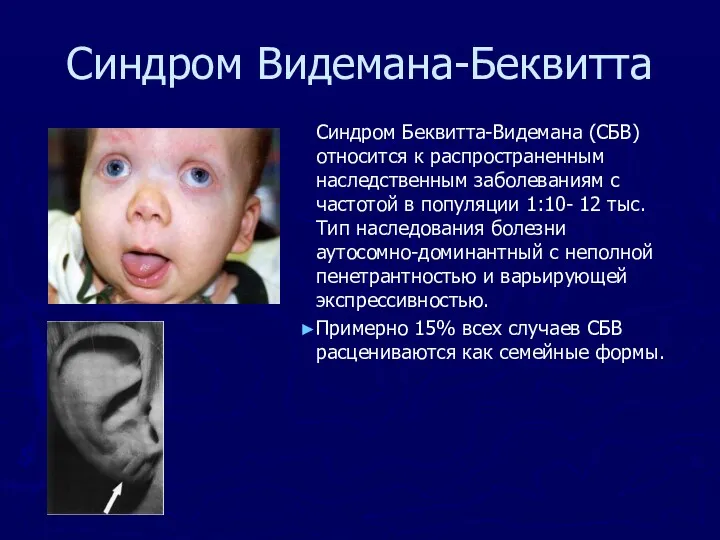
Синдром Беквитта-Видемана (СБВ) относится к распространенным наследственным заболеваниям с частотой
Синдром Видемана-Беквитта
Синдром Беквитта-Видемана (СБВ) относится к распространенным наследственным заболеваниям с частотой
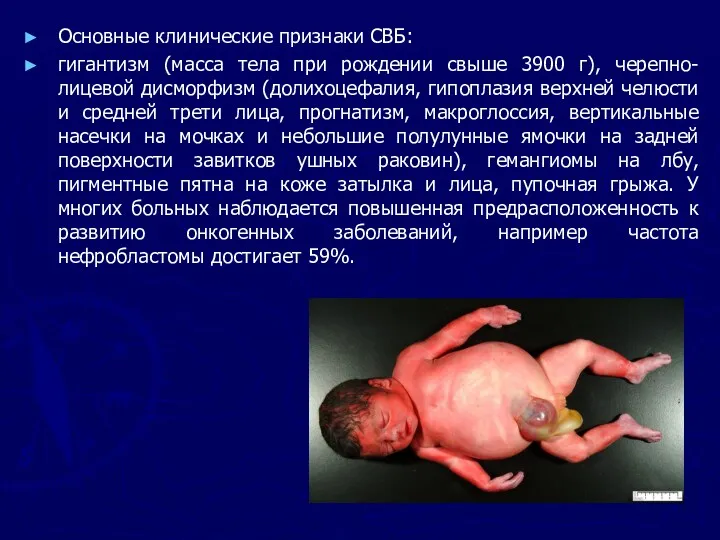
Основные клинические признаки СВБ:
гигантизм (масса тела при рождении свыше 3900
Основные клинические признаки СВБ:
гигантизм (масса тела при рождении свыше 3900

Генетика СВБ
Критической областью для СБВ является терминальная часть короткого плеча хромосомы
Генетика СВБ
Критической областью для СБВ является терминальная часть короткого плеча хромосомы

Генетика СВБ-
Преимущественная потеря материнских аллелей в опухолях, связанных с СВБ или
Преимущественная потеря материнских аллелей в опухолях, связанных с СВБ или

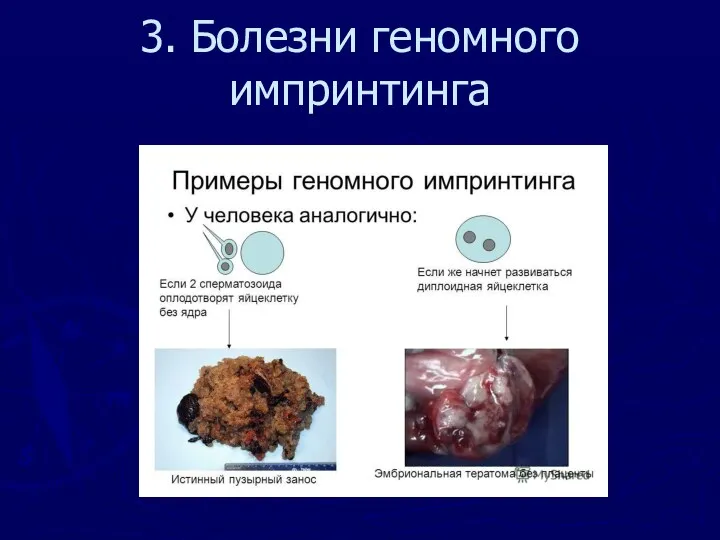
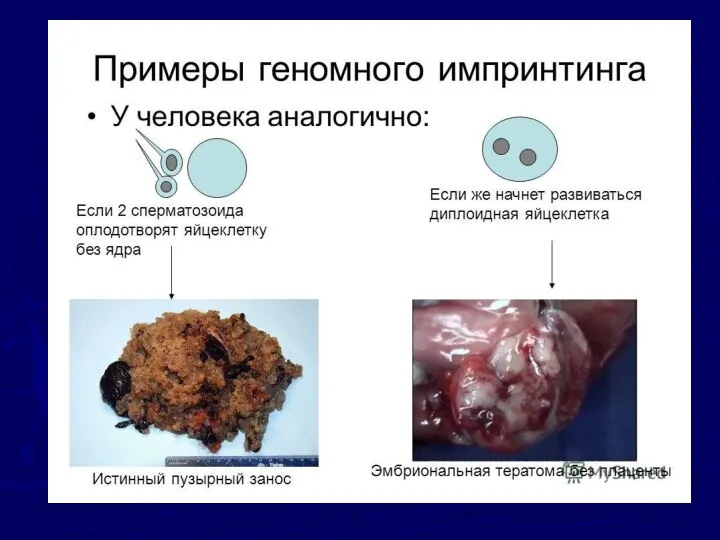
3. Болезни геномного импринтинга
3. Болезни геномного импринтинга


4. Болезни ошибок импринтинга
Болезни ошибок импринтинга - результат микроделеций в регуляторных
4. Болезни ошибок импринтинга
Болезни ошибок импринтинга - результат микроделеций в регуляторных
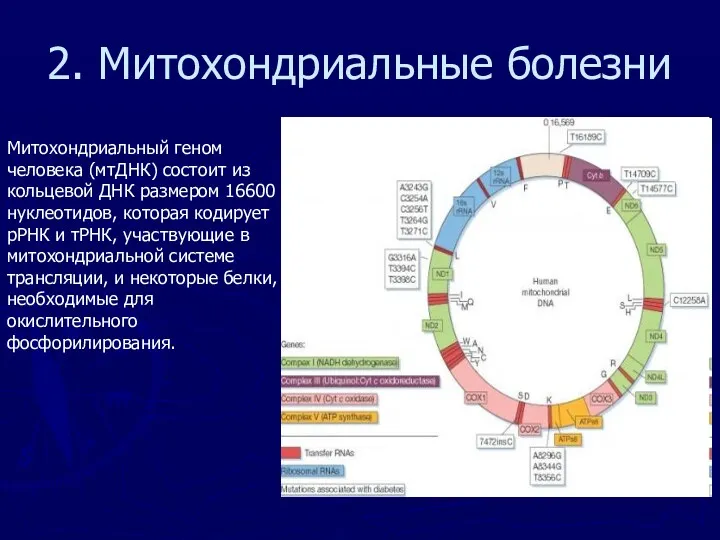
2. Митохондриальные болезни
Митохондриальный геном человека (мтДНК) состоит из кольцевой ДНК размером
2. Митохондриальные болезни
Митохондриальный геном человека (мтДНК) состоит из кольцевой ДНК размером

Митохондриальные заболевания
передаются только по женской линии, к детям обоих полов,
передаются только по женской линии, к детям обоих полов,

Можно выделить 2 группы митохондриальных заболеваний:
Ярко выраженные наследственные синдромы, обусловленные
Можно выделить 2 группы митохондриальных заболеваний:
Ярко выраженные наследственные синдромы, обусловленные

Синдром MELAS
(прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается
Синдром MELAS
(прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается

В каждом конкретном случае набор симптомов и их тяжесть может сильно
В каждом конкретном случае набор симптомов и их тяжесть может сильно
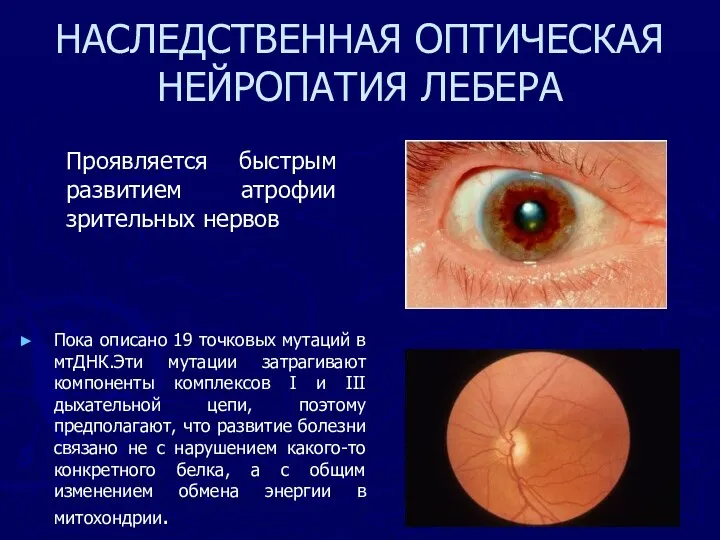
НАСЛЕДСТВЕННАЯ ОПТИЧЕСКАЯ НЕЙРОПАТИЯ ЛЕБЕРА
Пока описано 19 точковых мутаций в мтДНК.Эти мутации
НАСЛЕДСТВЕННАЯ ОПТИЧЕСКАЯ НЕЙРОПАТИЯ ЛЕБЕРА
Пока описано 19 точковых мутаций в мтДНК.Эти мутации
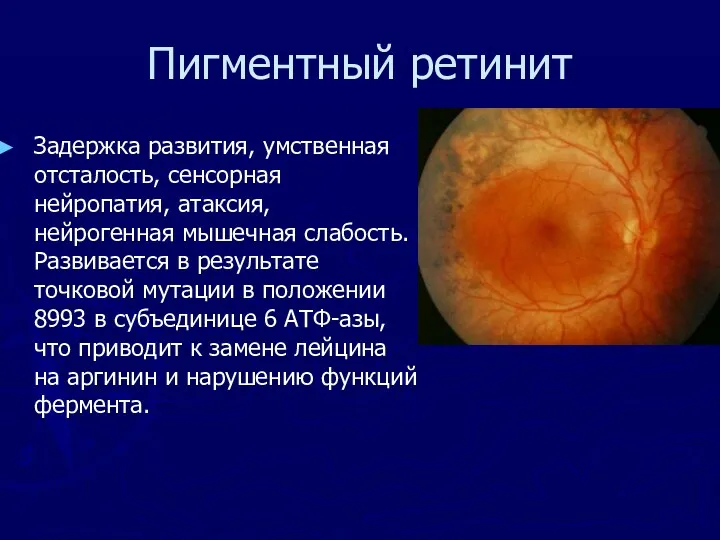
Пигментный ретинит
Задержка развития, умственная отсталость, сенсорная нейропатия, атаксия, нейрогенная мышечная слабость.
Пигментный ретинит
Задержка развития, умственная отсталость, сенсорная нейропатия, атаксия, нейрогенная мышечная слабость.

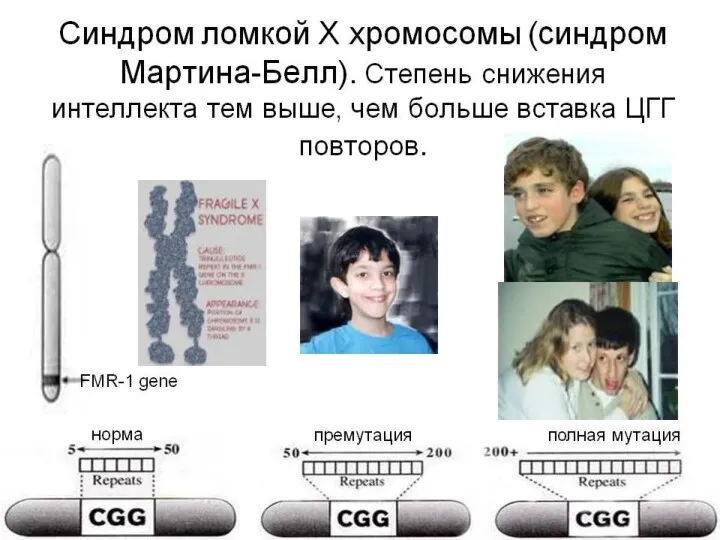
3. Болезни экспансии тринуклеотидных повторов (ЭТП)
Открытие феномена ЭТП было сделано при
3. Болезни экспансии тринуклеотидных повторов (ЭТП)
Открытие феномена ЭТП было сделано при
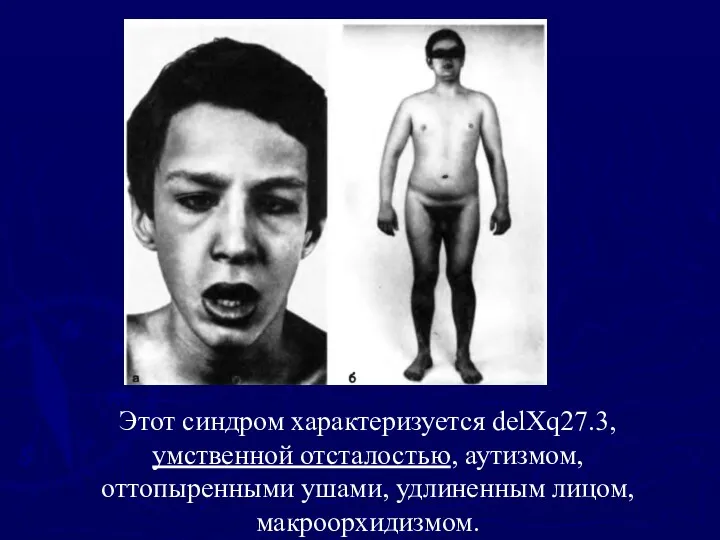
Этот синдром характеризуется delXq27.3,
умственной отсталостью, аутизмом,
оттопыренными ушами, удлиненным лицом, макроорхидизмом.
Этот синдром характеризуется delXq27.3,
умственной отсталостью, аутизмом,
оттопыренными ушами, удлиненным лицом, макроорхидизмом.

Необычность наследования данного синдрома заключается в том, что не все, а

В основе клинических проявления синдрома Мартина-Белла лежит многократное увеличение в первом
В основе клинических проявления синдрома Мартина-Белла лежит многократное увеличение в первом

Болезнь Гентингтона
- генетическое заболевание нервной системы, характеризующееся постепенным началом обычно
Болезнь Гентингтона
- генетическое заболевание нервной системы, характеризующееся постепенным началом обычно

Ген гентингтин (HTT)
кодирует белок гентингтин, расположен на коротком плече
Ген гентингтин (HTT)
кодирует белок гентингтин, расположен на коротком плече
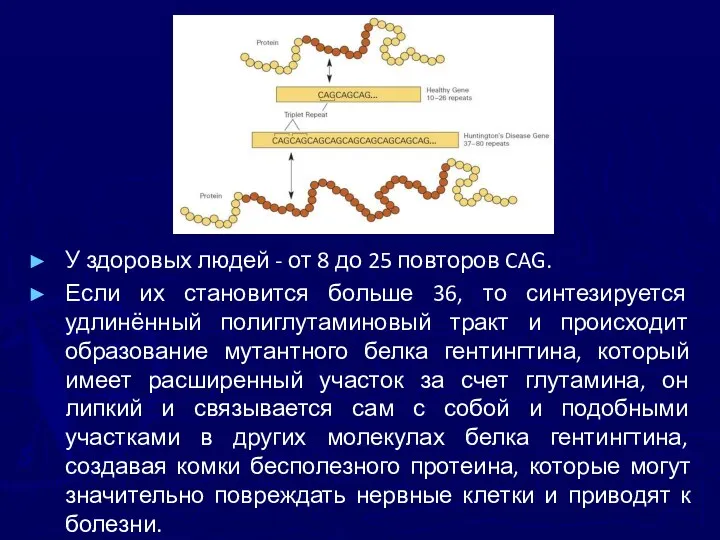
У здоровых людей - от 8 до 25 повторов CAG.
Если
У здоровых людей - от 8 до 25 повторов CAG.
Если

Болезнь Гентингтона поражает специфические области мозга.
Одними из первых бывают выразительные движения
Болезнь Гентингтона поражает специфические области мозга.
Одними из первых бывают выразительные движения

С момента появления первых симптомов продолжительность жизни составляет около 15–20 лет.
С момента появления первых симптомов продолжительность жизни составляет около 15–20 лет.
 Печень, строение и функции
Печень, строение и функции Движение. Значение движения. Движение растений. Органы движения животных. Виды скелетов
Движение. Значение движения. Движение растений. Органы движения животных. Виды скелетов Высшая нервная деятельность и ее возрастные особенности. Часть 1
Высшая нервная деятельность и ее возрастные особенности. Часть 1 Морфология человека. Конституция человека. Соматотипы
Морфология человека. Конституция человека. Соматотипы Возникновение жизни на Земле
Возникновение жизни на Земле Анатомия органов дыхания
Анатомия органов дыхания Викторина по биологии
Викторина по биологии презентация по биологии 8 класс
презентация по биологии 8 класс Закономірності впливу екологічних чинників на організми та їхні угруповання
Закономірності впливу екологічних чинників на організми та їхні угруповання Генетика микроорганизмов. Биотехнология. Генная инженерия
Генетика микроорганизмов. Биотехнология. Генная инженерия Внутренняя среда организма
Внутренняя среда организма Выращивание томата в теплице и парнике. 6 класс
Выращивание томата в теплице и парнике. 6 класс Надкласс рыбы. Игра
Надкласс рыбы. Игра Мир комнатных растений и их роль в жизни человека
Мир комнатных растений и их роль в жизни человека презентация и конспект урока урока
презентация и конспект урока урока Основные закономерности биологической эволюции
Основные закономерности биологической эволюции Мейоз
Мейоз Цианобактерии
Цианобактерии Царство грибов
Царство грибов Кити і дельфіни
Кити і дельфіни Свойства живых организмов
Свойства живых организмов Гемодинамика. Сердечно-сосудистая система и ее значение
Гемодинамика. Сердечно-сосудистая система и ее значение Класс Птицы
Класс Птицы Аксолотль. Места обитания
Аксолотль. Места обитания Зимующие птицы
Зимующие птицы Самые необычные кошки планеты
Самые необычные кошки планеты Центральная нервная система. Спинной мозг
Центральная нервная система. Спинной мозг Молекулярная биология. (Лекция 5-6)
Молекулярная биология. (Лекция 5-6)