- Біоінформатика. Парное выравнивание. (Тема 3)

Содержание
- 2. Парное выравнивание
- 3. При аналізі первинних структур процедура вирівнювання виявляє сходство між послідовностями (sequence similarity), яке може свідчити про
- 4. Гомологичные последовательности – последовательности, имеющие общее происхождение (общего предка). Признаки гомологичности белков сходная 3D-структура в той
- 5. Гомологи (?) Усе живе походить від одного загального предка, отже, усі послідовності є «гомологами». Насправді гомологи
- 6. Определение VLSPADKTNVKAAWAKVGAHAAGHG ||| | | |||| | |||| VLSEAEWQLVLHVWAKVEADVAGHG Выравнивание (alignment) – сравнение двух (парный) или
- 7. Что изображено? Название последовательности Номер столбца выравнивания Номер последнего в строке остатка ИЗ ЭТОЙ ПОСЛЕДОВАТЕЛЬНОСТИ Консервативный
- 8. «Идеальное» выравнивание – запись последовательностей одна под другой так, чтобы гомологичные фрагменты оказались друг под другом.
- 10. Какие задачи решает парное выравнивание? Нуклеотиды Изучение эволюционных связей Поиск генов, доменов, сигналов … Белки Изучение
- 11. Парное выравнивание – методы сравнения Глобальное выравнивание – находит лучшее решение для целых последовательностей. Локальное выравнивание
- 12. Информатика и Биоинформатика Біологічна задача Формалізація Алгоритм Алгоритм Алгоритм Алгоритм Алгоритм Тестування Параметры Параметры Параметри Параметры
- 13. Пример: сравнение последовательностей Тестирование: алгоритм должен распознавать последовательности, для которых известно, что они биологически (структурно и/или
- 14. Формалізація задачі через визначення редакційної відстані через визначення ваги вирівнювання.
- 15. Редакционное расстояние Элементарное преобразование последовательности: замена буквы или удаление буквы или вставка буквы. Редакционное расстояние: минимальное
- 16. Вага вирівнювання (alignment score) Якість співставлення двох послідовностей може бути описана за допомогою певного чисельного критерія.
- 17. Вычисление наилучшего выравнивания путем прохождения по Dot matrix для двух белков по 300 аминокислот требует 10^88
- 18. Парное выравнивание Человеческий гемоглобин (HH): VLSPADKTNVKAAWGKVGAHAGYEG Миоглобин кашалота (SWM): VLSEGEWQLVLHVWAKVEADVAGHG
- 19. Парное выравнивание - идентичность (HH) VLSPADKTNVKAAWGKVGAHAGYEG ||| | | || | | (SWM) VLSEGEWQLVLHVWAKVEADVAGHG Процент идентичности:
- 20. Парное выравнивание - сходство (HH) VLSPADKTNVKAAWGKVGAHAGYEG ||| . | | || | | (SWM) VLSEGEWQLVLHVWAKVEADVAGHG Процент
- 21. Парное выравнивание – вставка промежутков (gaps) (HH) VLSPADKTNVKAAWGKVGAH-AGYEG ⏐⏐⏐ . ⏐ ⏐ ⏐⏐ ⏐ ⏐⏐ ⏐
- 22. Парное выравнивание – вставка промежутков AKWTNLK----WAKV-ADVAGH-G ⏐⏐ ⏐⏐ ⏐ ⏐ ⏐⏐ ⏐ ⏐⏐⏐ ⏐ AK-TNVKAKLPWGKVGAHVAGEYG -
- 23. Парное выравнивание - Scoring (HH) VLSPADKTNVKAAWGKVGAH-AGYEG ||| . | | || || | (SWM) VLSEGEWQLVLHVWAKVEADVAGH-G Final
- 24. Парное выравнивание Алгоритмы парного выравнивания пробуют все возможные варианты выравнивания. Результат – выравнивание с наивысшей оценкой.
- 25. Система оценки - белки Идентичность: подсчитывается количество совпадений и делится на длину выравниваемого региона Similarity: Менее
- 26. Система оценки - белки Сходство: Положительная оценка для выравниваемых аминокислот из одной и той же группы.
- 27. Парное выравнивание Весовые матрицы (матрицы для оценки) – PAM, BLOSUM, Gonnet Системы оценки выравнивания различны для
- 28. Margaret Oakley Dayhoff 1972 год Сформулировала первую вероятностную модель эволюции белков
- 29. Матрицы сравнения белков Семейство матриц, которые отражают вероятность замены одной аминокислоты на другую во время эволюции.
- 30. PAM = Point Accepted Mutation Набор матриц, которые используются для выравнивания аминокислотных последовательностей белков Substitution Matrix
- 31. Еволюція терміна АРМ/РАМ зафіксовані (прийняті) точкові мутації (accepted point mutation), тобто амінокислотні заміни, що закріпилися в
- 32. PAM матрица PAM единицы отображают эволюционную дистанцию. 1 PAM единица – вероятность 1 точечной мутации на
- 33. PAM матрица PAM матрица базируется на последовательностях с 85% идентичности. У близких белков функции не должны
- 35. Относительная мутабельность аминокислот
- 36. Нормализованные частоты аминокислот
- 37. PAM 1 – матрица вероятностей
- 38. PAM 1 – нормализованная матрица вероятностей
- 39. PAM 250
- 40. PAM матрицы
- 41. Значення елементів вагової РАМ-матриці розраховується за формулою S(i,j) = 10 log10(Mij/pj) де S – вага співставлення
- 42. PAM 250 – весовая матрица
- 43. BLOSUM Matrices Blocks Substitution Matrices. Матрицы PAM обладают ограниченными возможностями, так как их «рейтинги замен» были
- 44. BLOSUM Блоки – короткие стабильные образы «шаблоны» по 3-60 aa длиной. Белки могут быть поделены на
- 45. BLOSUM62
- 48. Параметры по умолчанию Параметры для открытия\продления промежутков индивидуальны для каждой матрицы PAM30: open=9, extension=1 PAM250: open=14,
- 49. Параметры по умолчанию Выравнивания будут сильно отличаться при использовании различных параметров для промежутков. Для каждой матрицы
- 50. Параметры по умолчанию Мы можем использовать выравнвание последовательностей, базирующееся на структурном выравнивании. В этом случае структурное
- 51. Матрицы оценки DNA Похожесть нуклеотидов DNA определить невозможно. Основания делятся на 2 группы: пурины (A,G) и
- 52. Матрицы оценки DNA Мутации делятся на переходы (transitions) и превращения (transversions). Транзиции – пурин на пурин,
- 53. Матрицы оценки DNA De-facto транзиции происходят чаще.
- 54. Матрицы оценки DNA Унифицированная матрица подстановок нуклеотидов: Mismatch
- 55. Матрицы оценки DNA Неунифицированная матрица подстановок нуклеотидов:
- 56. Глобальное выравнивание Алгоритм Needleman and Wunsch (1970) Находит выравнивание двух полных последовательностей: ADLGAVFALCDRYFQ |||| |||| |
- 57. Локальное выравнивание Алгоритм Smith and Waterman (1981). Выполняет оптимальное выравнивание наиболее идентичного\похожего сегмента двух последовательностей. ADLGAVFALCDRYFQ
- 58. Выравнивание последовательностей методами динамического программирования Динамічне програмування – спосіб вирішення складних задач шляхом їх розбиття на
- 59. Выравнивание последовательностей методами динамического программирования У загальному випадку задача, що має оптимальну підструктуру, можна розв’язати за
- 60. Алгоритм Ніделмана-Вунша 1. Побудова ініціюючої матриці 2. Заповнення матриці 3. Пошук шляху вирівнювання
- 61. Алгоритм Ніделмана-Вунша 1. Побудова ініціюючої матриці
- 62. Дано: 2 последовательности x[1…n] и y[1…m] При сопоставлении x[1...i] и y[1…j] есть 3 варианта: Совпадение x[1…i-1]
- 63. Scoring matrix s(a,b), s(−, x) = s(x,−) = −d Fij – лучшая score-функция выравнивания x[1…i] and
- 64. Алгоритм Ніделмана-Вунша Заповнення матриці
- 65. Neddleman & Wunsch 1970 год Алгоритм: Начинает с конца последовательностей и продвигается, за каждый цикл сравнивая
- 66. Алгоритм Ніделмана-Вунша Пошук шляху вирівнювання
- 67. 13/02/2012 Маршрут выравнивания Needleman SB, Wunsch CD. A general method applicable to the search for similarities
- 68. 13/02/2012 Траектория, соответствующая оптимальному выравниванию
- 69. Важно: Выравнивание может не только окончиться, но и начаться в любом месте матрицы. Таким образом, вместо
- 70. Оценка Как можно оценить достоверность выравнивания? Какое выравнивание лучше ? ? Откуда взялись очки (оценка) :
- 71. Оценка неслучайности выравнивания Shuffle one of the sequences Align with the second sequence Calculate mean and
- 72. Данные с тем же набором, но с разным порядком: Перемешивание одной последовательности. Повтор выравнивания и его
- 73. Оценка неслучайности выравнивания x – вага вирівнювання двох вихідних послідовностей μ – усереднена вага отриманих у
- 75. Скачать презентацию

Парное выравнивание
Парное выравнивание

При аналізі первинних структур процедура вирівнювання виявляє сходство між послідовностями (sequence
При аналізі первинних структур процедура вирівнювання виявляє сходство між послідовностями (sequence

Гомологичные последовательности – последовательности, имеющие общее происхождение (общего предка).
Признаки гомологичности
Гомологичные последовательности – последовательности, имеющие общее происхождение (общего предка).
Признаки гомологичности

Гомологи (?)
Усе живе походить від одного загального предка, отже, усі послідовності
Гомологи (?)
Усе живе походить від одного загального предка, отже, усі послідовності
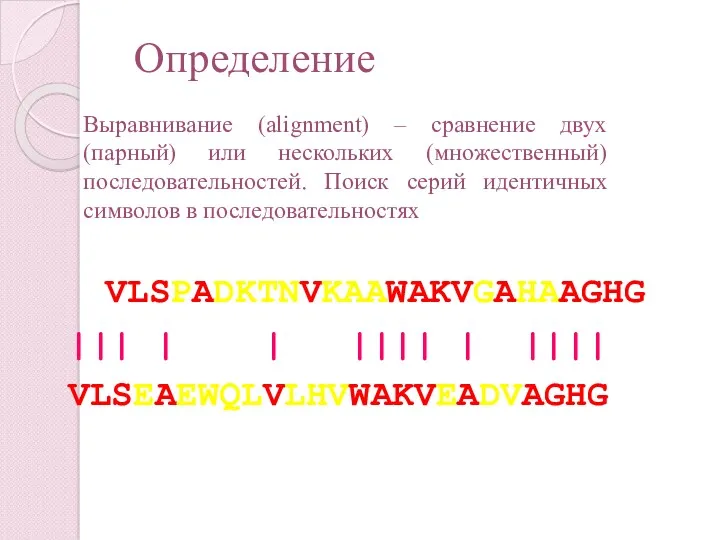
Определение
VLSPADKTNVKAAWAKVGAHAAGHG
||| | | |||| | ||||
VLSEAEWQLVLHVWAKVEADVAGHG
Выравнивание (alignment) – сравнение двух
Определение
VLSPADKTNVKAAWAKVGAHAAGHG
||| | | |||| | ||||
VLSEAEWQLVLHVWAKVEADVAGHG
Выравнивание (alignment) – сравнение двух
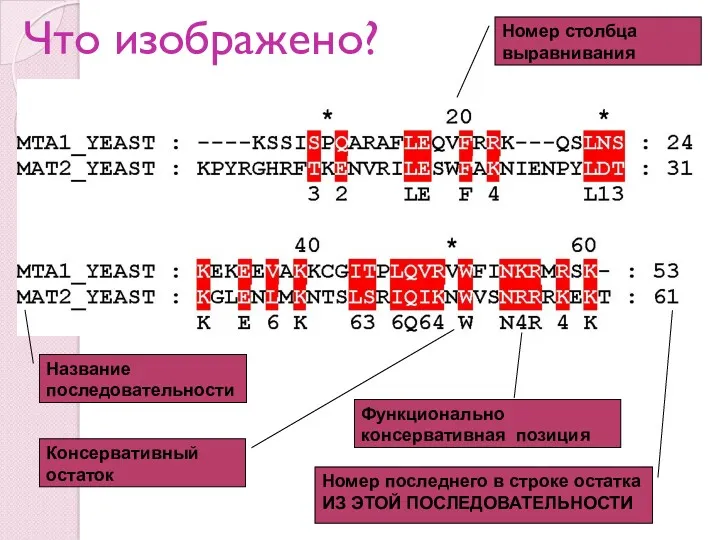
Что изображено?
Название последовательности
Номер столбца выравнивания
Номер последнего в строке остатка ИЗ ЭТОЙ
Что изображено?
Название последовательности
Номер столбца выравнивания
Номер последнего в строке остатка ИЗ ЭТОЙ

«Идеальное» выравнивание – запись последовательностей одна под другой так, чтобы гомологичные
«Идеальное» выравнивание – запись последовательностей одна под другой так, чтобы гомологичные

Какие задачи решает парное выравнивание?
Нуклеотиды
Изучение эволюционных связей
Поиск генов, доменов, сигналов …
Какие задачи решает парное выравнивание?
Нуклеотиды
Изучение эволюционных связей
Поиск генов, доменов, сигналов …
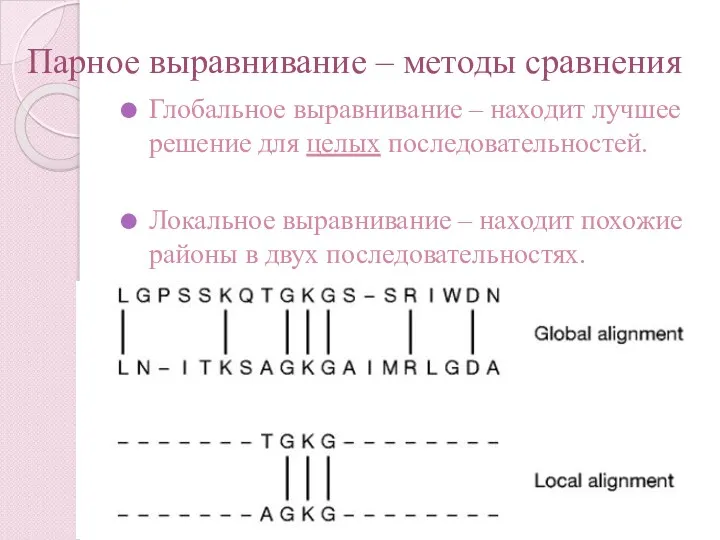
Парное выравнивание – методы сравнения
Глобальное выравнивание – находит лучшее решение для
Парное выравнивание – методы сравнения
Глобальное выравнивание – находит лучшее решение для
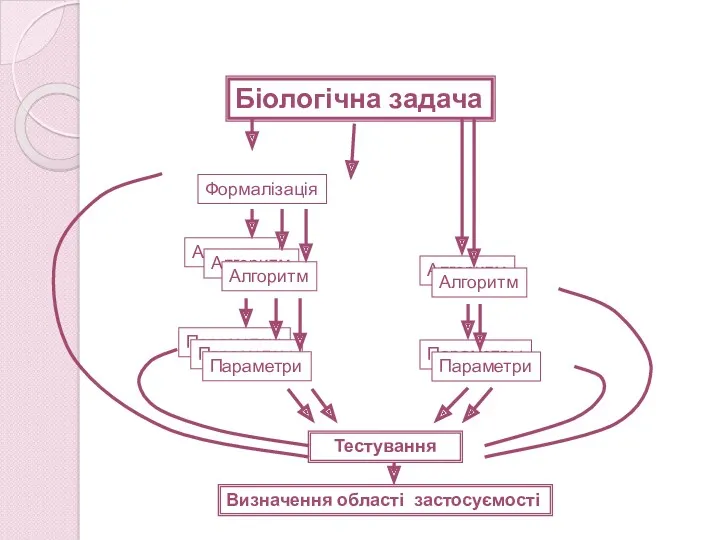
Информатика и Биоинформатика
Біологічна задача
Формалізація
Алгоритм
Алгоритм
Алгоритм
Алгоритм
Алгоритм
Тестування
Параметры
Параметры
Параметри
Параметры
Параметри
Визначення області застосуємості
Информатика и Биоинформатика
Біологічна задача
Формалізація
Алгоритм
Алгоритм
Алгоритм
Алгоритм
Алгоритм
Тестування
Параметры
Параметры
Параметри
Параметры
Параметри
Визначення області застосуємості

Пример: сравнение последовательностей
Тестирование: алгоритм должен распознавать последовательности, для которых известно, что
Пример: сравнение последовательностей
Тестирование: алгоритм должен распознавать последовательности, для которых известно, что

Формалізація задачі
через визначення редакційної відстані
через визначення ваги вирівнювання.
Формалізація задачі
через визначення редакційної відстані
через визначення ваги вирівнювання.

Редакционное расстояние
Элементарное преобразование последовательности: замена буквы или удаление буквы или вставка
Редакционное расстояние
Элементарное преобразование последовательности: замена буквы или удаление буквы или вставка

Вага вирівнювання (alignment score)
Якість співставлення двох послідовностей може бути описана за
Вага вирівнювання (alignment score)
Якість співставлення двох послідовностей може бути описана за

Вычисление наилучшего выравнивания путем прохождения по Dot matrix для двух белков
Вычисление наилучшего выравнивания путем прохождения по Dot matrix для двух белков

Парное выравнивание
Человеческий гемоглобин (HH):
VLSPADKTNVKAAWGKVGAHAGYEG
Миоглобин кашалота (SWM):
VLSEGEWQLVLHVWAKVEADVAGHG
Парное выравнивание
Человеческий гемоглобин (HH):
VLSPADKTNVKAAWGKVGAHAGYEG
Миоглобин кашалота (SWM):
VLSEGEWQLVLHVWAKVEADVAGHG

Парное выравнивание - идентичность
(HH) VLSPADKTNVKAAWGKVGAHAGYEG
||| | | || | |
(SWM)
Парное выравнивание - идентичность
(HH) VLSPADKTNVKAAWGKVGAHAGYEG
||| | | || | |
(SWM)

Парное выравнивание - сходство
(HH) VLSPADKTNVKAAWGKVGAHAGYEG
||| . | | || |
Парное выравнивание - сходство
(HH) VLSPADKTNVKAAWGKVGAHAGYEG
||| . | | || |

Парное выравнивание – вставка промежутков (gaps)
(HH) VLSPADKTNVKAAWGKVGAH-AGYEG
⏐⏐⏐ . ⏐ ⏐
Парное выравнивание – вставка промежутков (gaps)
(HH) VLSPADKTNVKAAWGKVGAH-AGYEG
⏐⏐⏐ . ⏐ ⏐

Парное выравнивание – вставка промежутков
AKWTNLK----WAKV-ADVAGH-G
⏐⏐ ⏐⏐ ⏐ ⏐ ⏐⏐ ⏐ ⏐⏐⏐
Парное выравнивание – вставка промежутков
AKWTNLK----WAKV-ADVAGH-G
⏐⏐ ⏐⏐ ⏐ ⏐ ⏐⏐ ⏐ ⏐⏐⏐

Парное выравнивание - Scoring
(HH) VLSPADKTNVKAAWGKVGAH-AGYEG
||| . | | || ||
Парное выравнивание - Scoring
(HH) VLSPADKTNVKAAWGKVGAH-AGYEG
||| . | | || ||

Парное выравнивание
Алгоритмы парного выравнивания пробуют все возможные варианты выравнивания.
Результат – выравнивание
Парное выравнивание
Алгоритмы парного выравнивания пробуют все возможные варианты выравнивания.
Результат – выравнивание

Система оценки - белки
Идентичность: подсчитывается количество совпадений и делится на длину
Система оценки - белки
Идентичность: подсчитывается количество совпадений и делится на длину
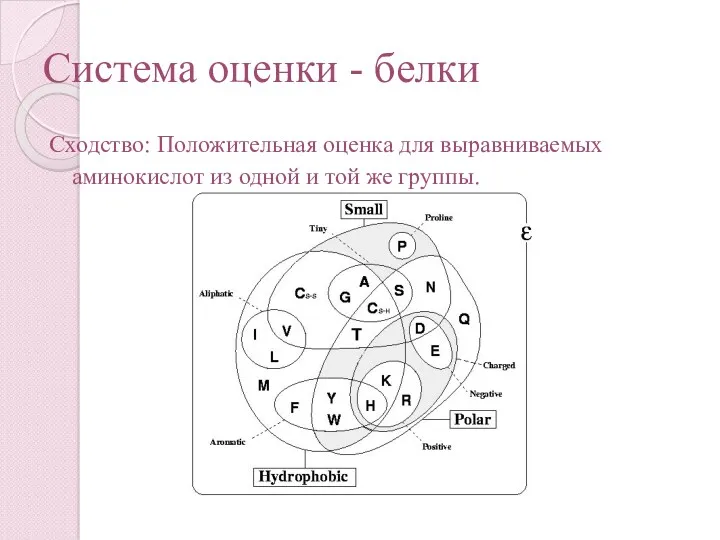
Система оценки - белки
Сходство: Положительная оценка для выравниваемых аминокислот из одной
Система оценки - белки
Сходство: Положительная оценка для выравниваемых аминокислот из одной

Парное выравнивание
Весовые матрицы (матрицы для оценки) – PAM, BLOSUM, Gonnet
Системы оценки
Парное выравнивание
Весовые матрицы (матрицы для оценки) – PAM, BLOSUM, Gonnet
Системы оценки

Margaret Oakley Dayhoff
1972 год
Сформулировала первую вероятностную модель эволюции белков
Margaret Oakley Dayhoff
1972 год
Сформулировала первую вероятностную модель эволюции белков

Матрицы сравнения белков
Семейство матриц, которые отражают вероятность замены одной аминокислоты на
Матрицы сравнения белков
Семейство матриц, которые отражают вероятность замены одной аминокислоты на

PAM = Point Accepted Mutation
Набор матриц, которые используются для выравнивания аминокислотных
PAM = Point Accepted Mutation
Набор матриц, которые используются для выравнивания аминокислотных

Еволюція терміна АРМ/РАМ
зафіксовані (прийняті) точкові мутації (accepted point mutation), тобто амінокислотні
Еволюція терміна АРМ/РАМ
зафіксовані (прийняті) точкові мутації (accepted point mutation), тобто амінокислотні

PAM матрица
PAM единицы отображают эволюционную дистанцию.
1 PAM единица – вероятность 1
PAM матрица
PAM единицы отображают эволюционную дистанцию.
1 PAM единица – вероятность 1

PAM матрица
PAM матрица базируется на последовательностях с 85% идентичности.
У близких белков
PAM матрица
PAM матрица базируется на последовательностях с 85% идентичности.
У близких белков

Относительная мутабельность аминокислот
Относительная мутабельность аминокислот
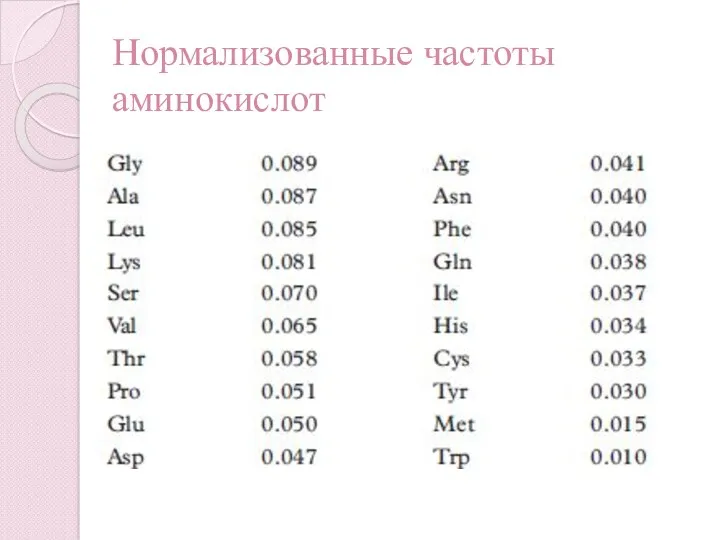
Нормализованные частоты аминокислот
Нормализованные частоты аминокислот
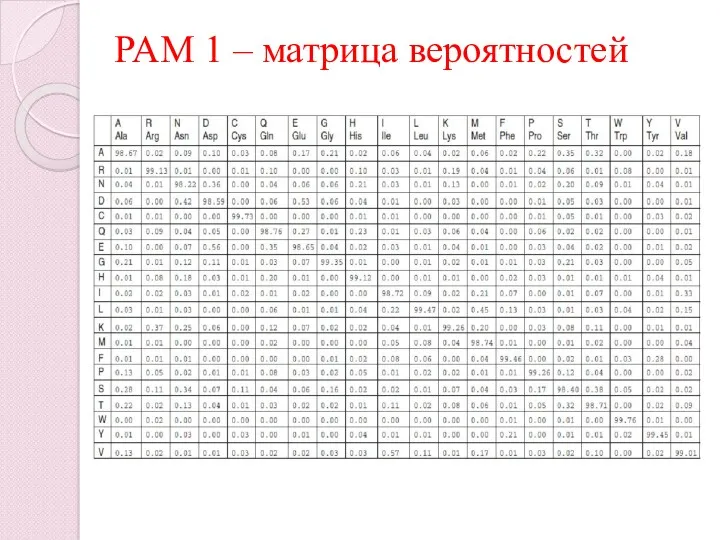
PAM 1 – матрица вероятностей
PAM 1 – матрица вероятностей

PAM 1 – нормализованная матрица вероятностей
PAM 1 – нормализованная матрица вероятностей

PAM 250
PAM 250
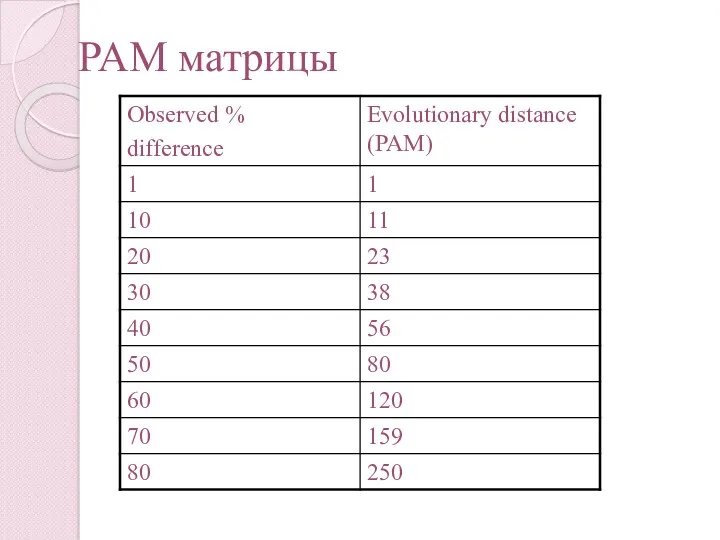
PAM матрицы
PAM матрицы

Значення елементів вагової РАМ-матриці розраховується за формулою
S(i,j) = 10 log10(Mij/pj)
де S
Значення елементів вагової РАМ-матриці розраховується за формулою
S(i,j) = 10 log10(Mij/pj)
де S
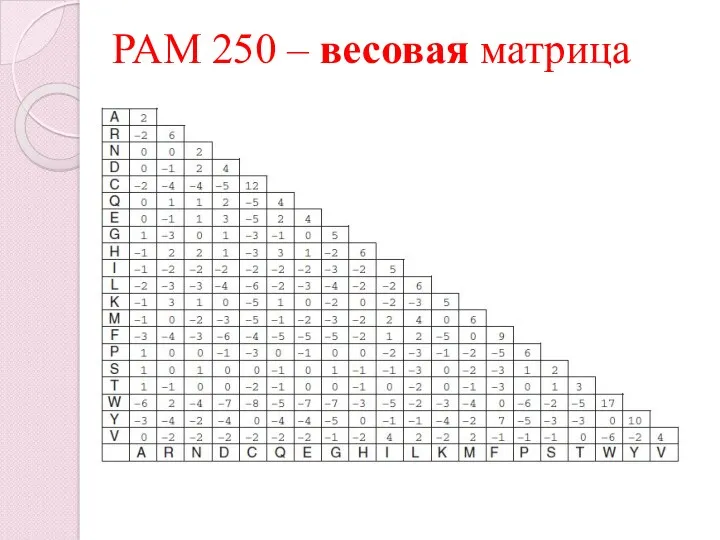
PAM 250 – весовая матрица
PAM 250 – весовая матрица

BLOSUM Matrices
Blocks Substitution Matrices.
Матрицы PAM обладают ограниченными возможностями, так как их
BLOSUM Matrices
Blocks Substitution Matrices.
Матрицы PAM обладают ограниченными возможностями, так как их

BLOSUM
Блоки – короткие стабильные образы «шаблоны» по 3-60 aa длиной.
Белки могут
BLOSUM
Блоки – короткие стабильные образы «шаблоны» по 3-60 aa длиной.
Белки могут
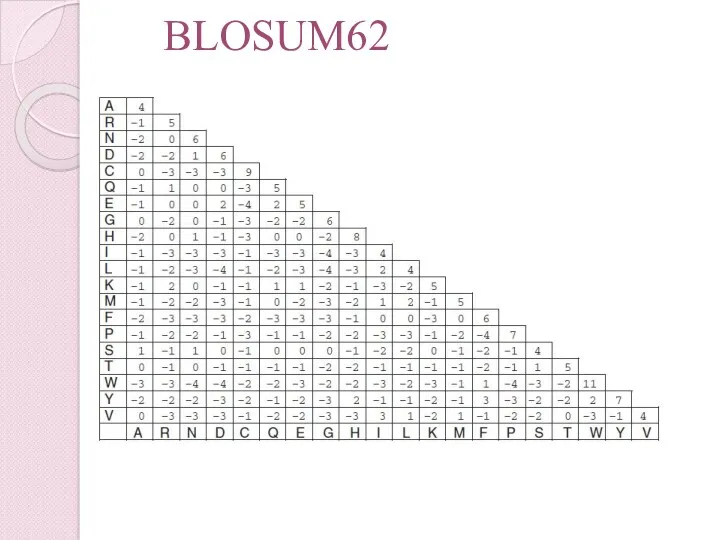
BLOSUM62
BLOSUM62
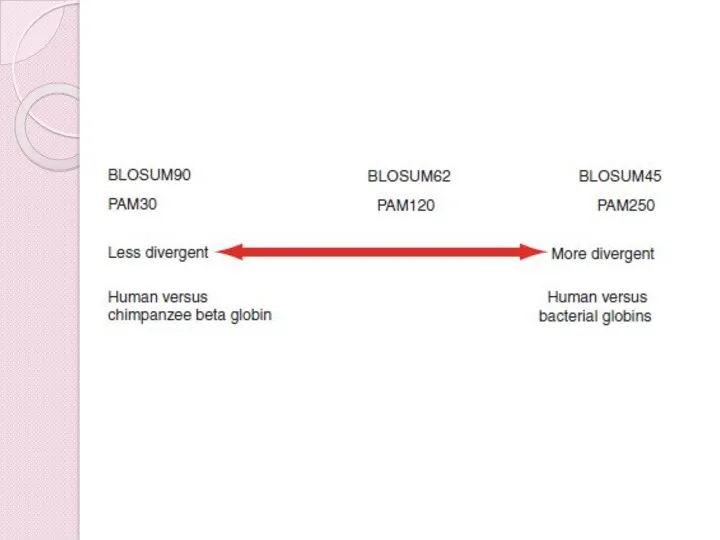

Параметры по умолчанию
Параметры для открытия\продления промежутков индивидуальны для каждой матрицы
PAM30: open=9,
Параметры по умолчанию
Параметры для открытия\продления промежутков индивидуальны для каждой матрицы
PAM30: open=9,

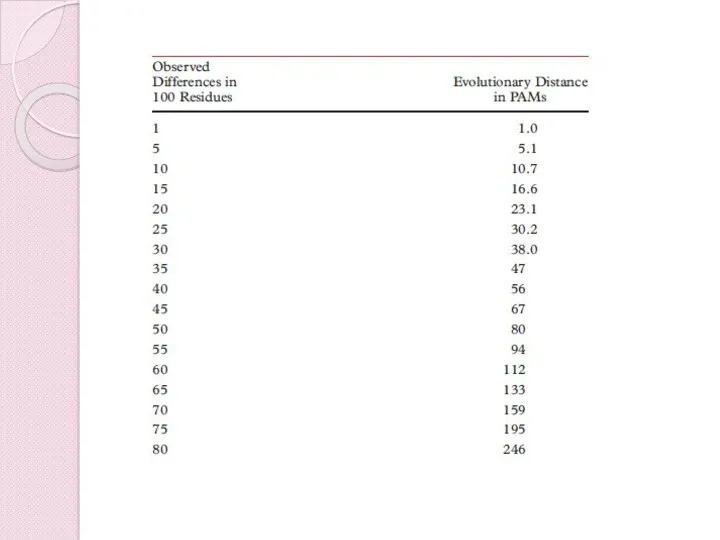
Параметры по умолчанию
Выравнивания будут сильно отличаться при использовании различных параметров для
Параметры по умолчанию
Выравнивания будут сильно отличаться при использовании различных параметров для
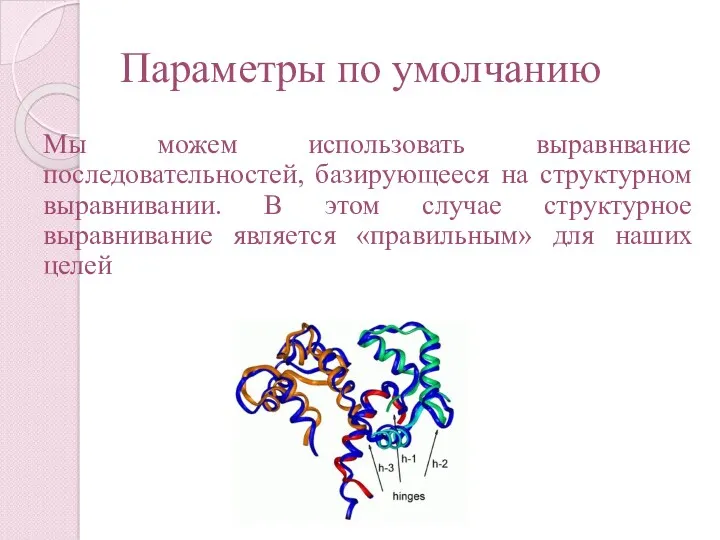
Параметры по умолчанию
Мы можем использовать выравнвание последовательностей, базирующееся на структурном выравнивании.
Параметры по умолчанию
Мы можем использовать выравнвание последовательностей, базирующееся на структурном выравнивании.
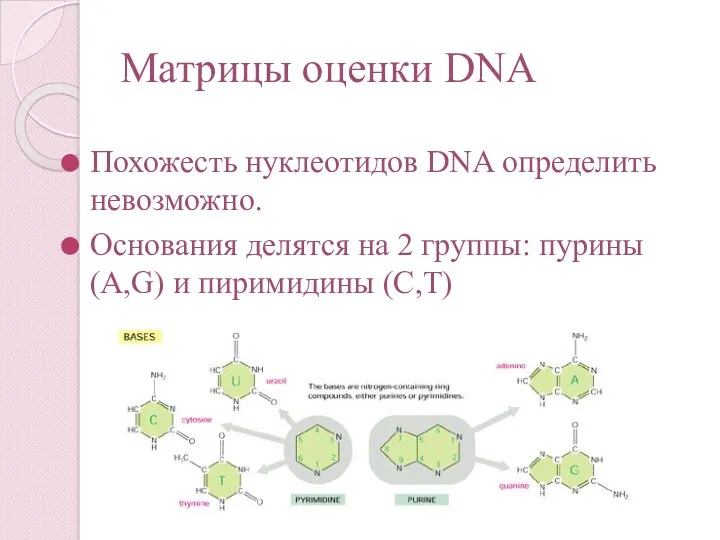
Матрицы оценки DNA
Похожесть нуклеотидов DNA определить невозможно.
Основания делятся на 2 группы:
Матрицы оценки DNA
Похожесть нуклеотидов DNA определить невозможно.
Основания делятся на 2 группы:

Матрицы оценки DNA
Мутации делятся на переходы (transitions) и превращения (transversions).
Транзиции
Матрицы оценки DNA
Мутации делятся на переходы (transitions) и превращения (transversions).
Транзиции

Матрицы оценки DNA
De-facto транзиции происходят чаще.
Матрицы оценки DNA
De-facto транзиции происходят чаще.
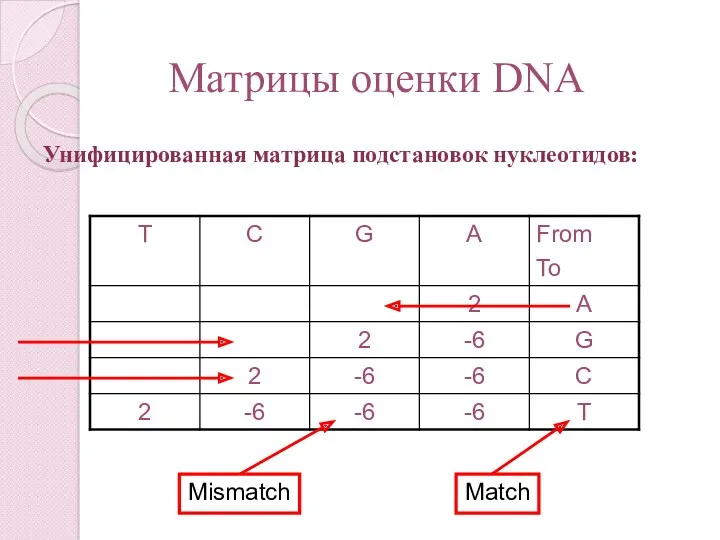
Матрицы оценки DNA
Унифицированная матрица подстановок нуклеотидов:
Mismatch
Матрицы оценки DNA
Унифицированная матрица подстановок нуклеотидов:
Mismatch

Матрицы оценки DNA
Неунифицированная матрица подстановок нуклеотидов:
Матрицы оценки DNA
Неунифицированная матрица подстановок нуклеотидов:

Глобальное выравнивание
Алгоритм Needleman and Wunsch (1970)
Находит выравнивание двух полных последовательностей:
Глобальное выравнивание
Алгоритм Needleman and Wunsch (1970)
Находит выравнивание двух полных последовательностей:
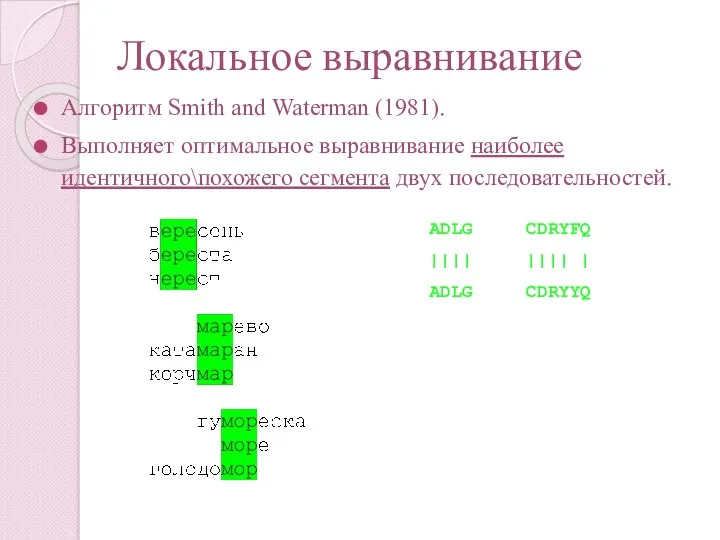
Локальное выравнивание
Алгоритм Smith and Waterman (1981).
Выполняет оптимальное выравнивание наиболее идентичного\похожего сегмента
Локальное выравнивание
Алгоритм Smith and Waterman (1981).
Выполняет оптимальное выравнивание наиболее идентичного\похожего сегмента

Выравнивание последовательностей методами динамического программирования
Динамічне програмування – спосіб вирішення складних задач
Выравнивание последовательностей методами динамического программирования
Динамічне програмування – спосіб вирішення складних задач

Выравнивание последовательностей методами динамического программирования
У загальному випадку задача, що має оптимальну
Выравнивание последовательностей методами динамического программирования
У загальному випадку задача, що має оптимальну

Алгоритм Ніделмана-Вунша
1. Побудова ініціюючої матриці
2. Заповнення матриці
3. Пошук шляху вирівнювання
Алгоритм Ніделмана-Вунша
1. Побудова ініціюючої матриці
2. Заповнення матриці
3. Пошук шляху вирівнювання
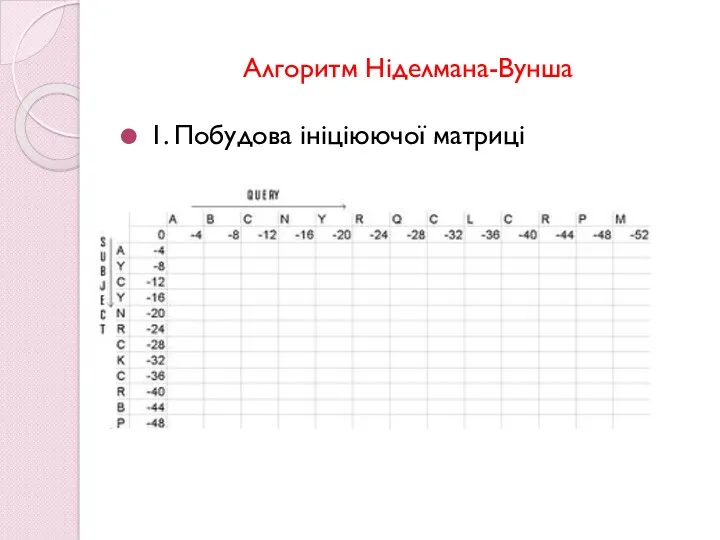
Алгоритм Ніделмана-Вунша
1. Побудова ініціюючої матриці
Алгоритм Ніделмана-Вунша
1. Побудова ініціюючої матриці
![Дано: 2 последовательности x[1…n] и y[1…m] При сопоставлении x[1...i] и](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/235530/slide-61.jpg)
Дано: 2 последовательности x[1…n] и y[1…m]
При сопоставлении x[1...i] и y[1…j]
Дано: 2 последовательности x[1…n] и y[1…m]
При сопоставлении x[1...i] и y[1…j]

Scoring matrix s(a,b), s(−, x) = s(x,−) = −d
Fij – лучшая
Scoring matrix s(a,b), s(−, x) = s(x,−) = −d
Fij – лучшая

Алгоритм Ніделмана-Вунша
Заповнення матриці
Алгоритм Ніделмана-Вунша
Заповнення матриці

Neddleman & Wunsch
1970 год
Алгоритм:
Начинает с конца последовательностей и продвигается, за каждый
Neddleman & Wunsch
1970 год
Алгоритм:
Начинает с конца последовательностей и продвигается, за каждый
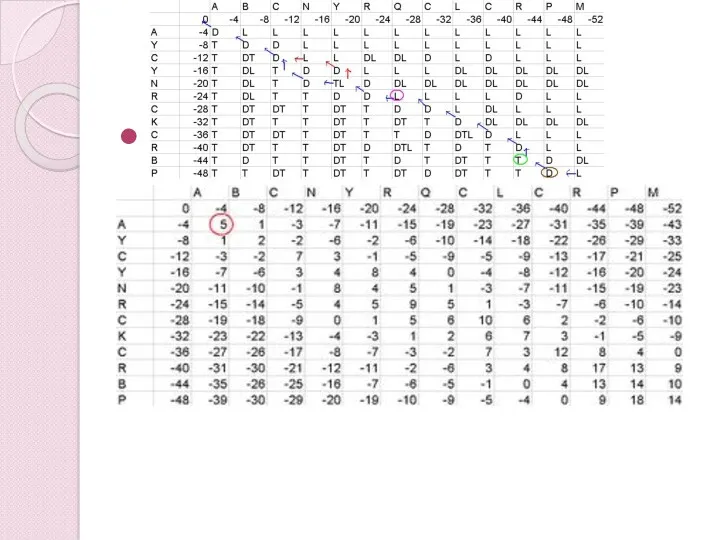
Алгоритм Ніделмана-Вунша
Пошук шляху вирівнювання
Алгоритм Ніделмана-Вунша
Пошук шляху вирівнювання
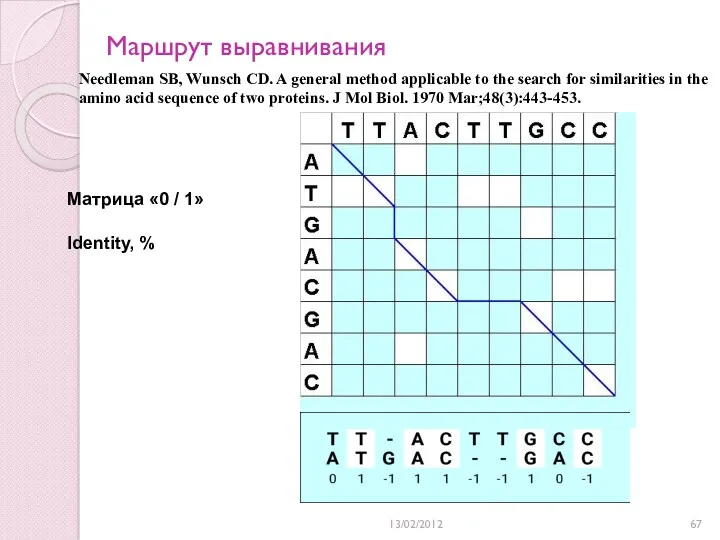
13/02/2012
Маршрут выравнивания
Needleman SB, Wunsch CD. A general method applicable to the
13/02/2012
Маршрут выравнивания
Needleman SB, Wunsch CD. A general method applicable to the
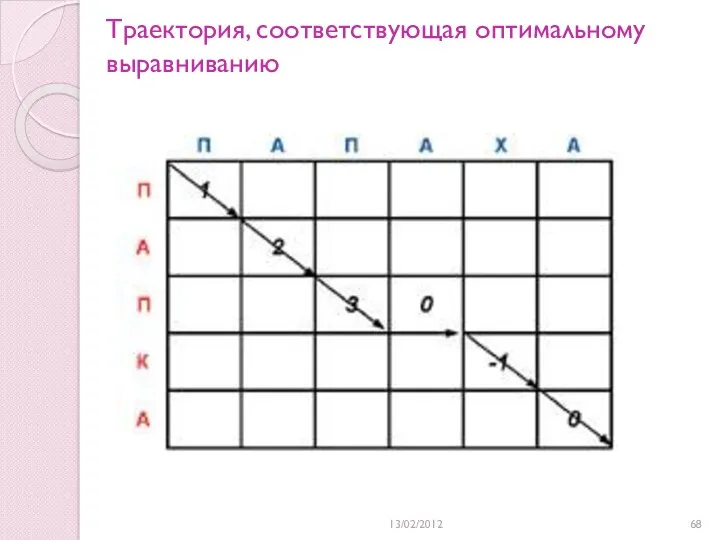
13/02/2012
Траектория, соответствующая оптимальному выравниванию
13/02/2012
Траектория, соответствующая оптимальному выравниванию
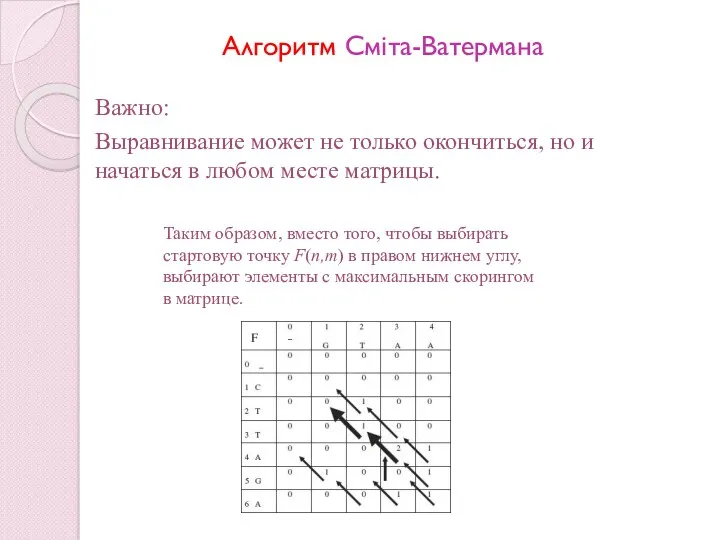
Важно:
Выравнивание может не только окончиться, но и начаться в любом месте
Важно:
Выравнивание может не только окончиться, но и начаться в любом месте

Оценка
Как можно оценить достоверность выравнивания?
Какое выравнивание лучше ?
?
Откуда взялись очки (оценка)
Оценка
Как можно оценить достоверность выравнивания?
Какое выравнивание лучше ?
?
Откуда взялись очки (оценка)
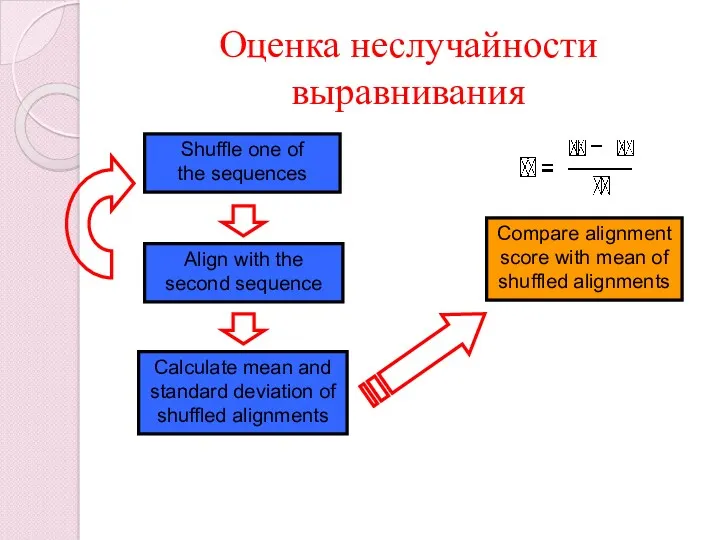
Оценка неслучайности выравнивания
Shuffle one of
the sequences
Align with the
second sequence
Calculate
Оценка неслучайности выравнивания
Shuffle one of
the sequences
Align with the
second sequence
Calculate

Данные с тем же набором, но с разным порядком:
Перемешивание одной последовательности.
Повтор
Данные с тем же набором, но с разным порядком:
Перемешивание одной последовательности.
Повтор

Оценка неслучайности выравнивания
x – вага вирівнювання двох вихідних послідовностей
μ –
Оценка неслучайности выравнивания
x – вага вирівнювання двох вихідних послідовностей
μ –
 Микробиологиямен танысу
Микробиологиямен танысу Прокариоты. Уровни клеточной организации
Прокариоты. Уровни клеточной организации Законы Г. Менделя
Законы Г. Менделя Приспособленность – результат действия факторов эволюции
Приспособленность – результат действия факторов эволюции Мышечная система
Мышечная система Листья и плоды деревьев и кустарников
Листья и плоды деревьев и кустарников Вирустардың таксономиясы
Вирустардың таксономиясы Умови плавання тіл. (Бінарний урок. 7 клас)
Умови плавання тіл. (Бінарний урок. 7 клас) Класс насекомые
Класс насекомые Результаты эволюции: приспособленность организмов и многообразие видов
Результаты эволюции: приспособленность организмов и многообразие видов Класс Ракообразные
Класс Ракообразные Обмен веществ - основа существования клетки
Обмен веществ - основа существования клетки Регуляция функций в организме
Регуляция функций в организме Чибис - птица 2010 года
Чибис - птица 2010 года Функциональная анатомия мышц конечностей
Функциональная анатомия мышц конечностей Клеточная оболочка и цитоплазма
Клеточная оболочка и цитоплазма Функции популяции
Функции популяции Биологические ресурсы мира
Биологические ресурсы мира ЕГЭ на 22 год и анализ 21 года
ЕГЭ на 22 год и анализ 21 года Человек и природа. Красная и черная книги
Человек и природа. Красная и черная книги Органы чувств. Анализаторы
Органы чувств. Анализаторы Таргетинг генов
Таргетинг генов Фитопатогенные нематоды. (Лекция 8)
Фитопатогенные нематоды. (Лекция 8) Биоиндикация на разных уpовнях организации живого
Биоиндикация на разных уpовнях организации живого Как появился человек на Земле
Как появился человек на Земле Класс Млекопитающие. Семейство: Парнокопытные и Непарнокопытные. 7 класс
Класс Млекопитающие. Семейство: Парнокопытные и Непарнокопытные. 7 класс Eukaryotic cell structure
Eukaryotic cell structure Органы дыхания. Эволюция
Органы дыхания. Эволюция