- Кислотность почв

Содержание
- 2. Кислотность почв КИСЛОТНОСТЬ ПОЧВ – способность почвы проявлять свойства кислот. Мера кислотности почв – концентрация (точнее
- 3. Кислотность почв pH = – lg [H+] = – lg [H3O+] Водородный показатель (рН) – величина,
- 4. Кислотность почв По предложению датского физико-химика и биохимика Сёрена Петера Лаурица Сёренсена вместо значений [H+] используют
- 5. Кислотность почв Величину рН используют как меру кислотности, нейтральности или основности водных растворов: в кислых растворах
- 6. Кислотность почв Кислотность почвы актуальная или активная – кислотность почвенного раствора, почвенной суспензии или водной вытяжки
- 7. Кислотность почв Кислотность почвы потенциальная или пассивная – кислотность твёрдой фазы почвы, обусловленная ионами гидроксония (т.
- 8. Кислотность почв Кислотность почвы обменная – кислотность, вызываемая присутствием в поглощенном состоянии ионов гидроксония (т. н.
- 9. Образование ДЭС-мицеллы
- 10. Строение ДЭС-мицеллы
- 11. Кислотность почв Кислотность почвы гидролитическая – кислотность, обусловлена наличием в почвенном поглощающем комплексе ионов гидроксония (т.
- 12. Кислотность почв
- 13. Кислотность почв В агрохимии в зависимости от величины pH почвы подразделяются:
- 14. Кислотность почв и растения Основываясь на многочисленных экспериментальных данных, показывающих зависимость распространения отдельных видов растений и
- 15. Кислотность почв и растения Жозиас Браун-Бланке ‒ один из крупнейших геоботаников XX века, основатель и руководитель
- 16. Кислотность почв и растения Михаил Владимирович Марков считал, что растения и растительные сообщества, связанные с резкощелоч-ными
- 17. Кислотность почв и растения На основании своих наблюдений и критического анализа литературных источников М. В. Марков
- 18. Теории кислот и оснований Существует 3 основных теории кислот и оснований ‒ их создали Аррениус и
- 19. Теория Аррениуса-Оствальда Сванте Август Аррениус – шведский физико-химик, автор теории электролитической диссоциации, лауреат Нобелевской премии по
- 20. Теория Аррениуса-Оствальда Согласно теории Сванте Августа Аррениуса и Вильгельма Фридриха Оствальда Кислоты – вещества, при электролитической
- 21. Теория Брёнстеда-Лоури Йоханнес-Николаус Брёнстед – датский физикохимик, и Томас Мартин Лаури (Лоури) – британский химик –
- 22. Теория Брёнстеда-Лоури Согласно протонной теории (теории сопряженных кислот-оснований), кислота – соединение, способное отдавать основанию катионы водорода
- 23. Теория Брёнстеда-Лоури При взаимодействии кислоты и основания, кислота отдаёт свои протоны, а основание их принимает (связывает).
- 24. Кислотность почв рН играет существенную роль в реакциях перехода протонов (в реакциях брёнстедовых кислот и оснований).
- 25. Кислотность почв БРЁНСТЕДОВЫ КИСЛОТЫ − нейтральные молекулы или ионы, способные предоставлять протон другой молекуле или иону
- 26. Теория кислот и оснований Льюиса Гилберт Ньютон Льюис – американский физикохимик, предложивший и развивший (1912-1916) электронную
- 27. Теория кислот и оснований Льюиса В теории Гилберта Ньютона Льюиса (Электронной Теории Кислот-Оснований) было ещё более
- 28. Теория кислот и оснований Льюиса Кислоты по Льюису: 1) любые химические соединения, которые возбуждают реакцию свободной
- 29. Теория кислот и оснований Льюиса Электронные орбитали могут быть молекулярными орбитами, связями или атомными орбиталями в
- 30. Теория кислот и оснований Льюиса ЛЬЮИСОВЫ КИСЛОТЫ ЖЁСТКИЕ (СИЛЬНЫЕ) ‒ 1) молекулярная частица сравнительно небольшого размера,
- 31. Теория кислот и оснований Льюиса Внутри группы жёстких (сильных) льюисовых кислот существует не единый уровень “жёсткости”
- 32. Теория кислот и оснований Льюиса Оценить силу льюисовских кислот можно по параметру Мисоно. Обычно сильные льюисовские
- 33. Теория кислот и оснований Льюиса ЛЬЮИСОВЫ КИСЛОТЫ МЯГКИЕ (СЛАБЫЕ) ‒ 1) частицы сравнительно большого размера, слабо-окисленные,
- 34. Теория кислот и оснований Льюиса Примерами мягких (слабых) кислот по Льюису служат Cd2+, Сu+, Hg+ и
- 35. Теория кислот и оснований Льюиса Список сильных и слабых кислот по Льюису, присутствующих в почвенных растворах.
- 36. Теория кислот и оснований Льюиса ЛЬЮИСОВЫ ОСНОВАНИЯ ‒ 1) соединения, возбуждающие реакцию дважды населенной электронной орбиталью;
- 37. Теория кислот и оснований Льюиса К льюисовым основаниям относятся Н2О, оксианионы типа ОН−, СОО−, СО32−, SO42−
- 38. Теория кислот и оснований Льюиса ЛЬЮИСОВЫ ОСНОВАНИЯ ЖЁСТКИЕ (СИЛЬНЫЕ) − 1) сильно электроотрицательная и слабополяризуемая молекулярная
- 39. Теория кислот и оснований Льюиса В жёстких (сильных) льюисовых основаниях электронные облака расположены ближе к ядру
- 40. Теория кислот и оснований Льюиса ЛЬЮИСОВЫ ОСНОВАНИЯ МЯГКИЕ (СЛАБЫЕ) ‒ частицы с электронодонорными атомами, обладающими низкой
- 41. Теория кислот и оснований Льюиса Список сильных и слабых оснований по Льюису, присутствующих в почвенных растворах.
- 42. Сопоставление теорий кислот и оснований *неподелённые пары электронов
- 43. Буферность почв Буферность почвы − способность почвы противостоять изменению её свойств при воздействии различных факторов [ГОСТ
- 44. Буферность почв Буферность почвы кислотно-щелочная – способность жидкой и твёрдой фаз почвы противостоять изменению реакции среды
- 45. Буферность почв Высокая буферность проявляется в тех случаях, когда концентрации компонентов буферного раствора значительно превосходят вводимые
- 46. Буферность почв Уравнение Гендерсона–Хассельбаха – логарифмическая форма уравнения константы равновесия диссоциации слабых кислот Кa: pH =
- 47. Буферность почв Аналогичное уравнение может быть выведено и для щелочных растворов: , где BOH – основание
- 48. Буферность почв Буферная ёмкость – количество сильной кислоты (или щелочи), которое надо прибавить к буферной системе,
- 49. Буферность почв Буферные кислотно-щелочные системы – системы способные противостоять изменению рН при подкислении или подщелачивании. Кислотно-щелочные
- 50. Буферность почв Свойства буферных кислотно-щелочных систем, содержащих слабую кислоту (или слабое основание) и её соль, количественно
- 51. Буферность почв Если буферная кислотно-щелочная система представлена слабым основанием и его солью, то величина рОН =
- 52. Буферность почв Примеры буферных кислотно-щелочных систем: а) ацетатной буферной смесь: СН3СООН – Н2О – СН3СООNa, для
- 53. Буферность почв Буферные кислотно-щелочные системы, как известно, обладают способностью поддерживать рН на относительно постоянном уровне; добавление
- 54. Механизм кислотно-основной буферности почв (по Ульриху)
- 55. Буферность почв Изотоническая буферность почв − буферность, позволяющая поддерживать постоянное осмотическое давление в почве Изотонические растворы
- 56. Буферность почв Окислительно-восстановительная буферность почв − буферность, позволяющая поддерживать окислительно-восстановительный потенциал. Окислительно-восстановительный потенциал − потенциал, устанавливающийся
- 58. Скачать презентацию

Кислотность почв
КИСЛОТНОСТЬ ПОЧВ – способность почвы проявлять свойства кислот.
Мера кислотности почв
Кислотность почв
КИСЛОТНОСТЬ ПОЧВ – способность почвы проявлять свойства кислот.
Мера кислотности почв
![Кислотность почв pH = – lg [H+] = – lg](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/165837/slide-2.jpg)
Кислотность почв
pH = – lg [H+] = – lg [H3O+]
Водородный
Кислотность почв
pH = – lg [H+] = – lg [H3O+]
Водородный

Кислотность почв
По предложению датского физико-химика и биохимика Сёрена Петера Лаурица Сёренсена
Кислотность почв
По предложению датского физико-химика и биохимика Сёрена Петера Лаурица Сёренсена

Кислотность почв
Величину рН используют как меру кислотности, нейтральности или основности водных
Кислотность почв
Величину рН используют как меру кислотности, нейтральности или основности водных

Кислотность почв
Кислотность почвы актуальная или активная – кислотность почвенного раствора, почвенной
Кислотность почв
Кислотность почвы актуальная или активная – кислотность почвенного раствора, почвенной

Кислотность почв
Кислотность почвы потенциальная или пассивная – кислотность твёрдой фазы почвы,
Кислотность почв
Кислотность почвы потенциальная или пассивная – кислотность твёрдой фазы почвы,

Кислотность почв
Кислотность почвы обменная – кислотность, вызываемая присутствием в поглощенном состоянии
Кислотность почв
Кислотность почвы обменная – кислотность, вызываемая присутствием в поглощенном состоянии
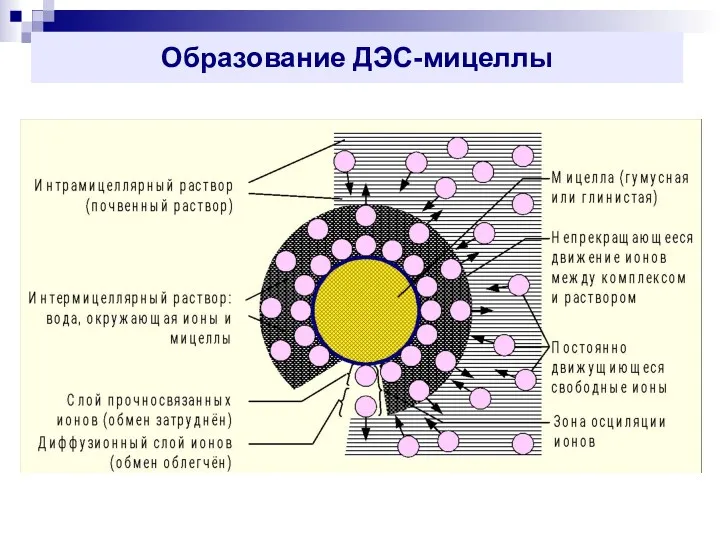
Образование ДЭС-мицеллы
Образование ДЭС-мицеллы

Строение ДЭС-мицеллы
Строение ДЭС-мицеллы

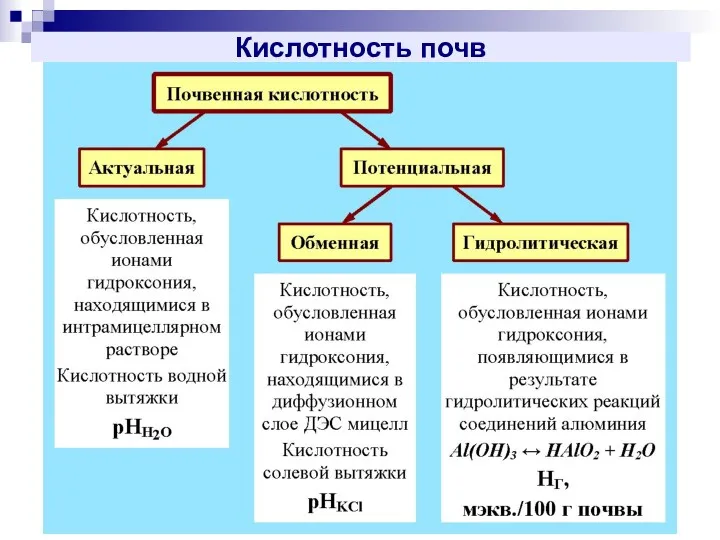
Кислотность почв
Кислотность почвы гидролитическая – кислотность, обусловлена наличием в почвенном поглощающем
Кислотность почв
Кислотность почвы гидролитическая – кислотность, обусловлена наличием в почвенном поглощающем
Кислотность почв
Кислотность почв

Кислотность почв
В агрохимии в зависимости от величины pH почвы подразделяются:
Кислотность почв
В агрохимии в зависимости от величины pH почвы подразделяются:

Кислотность почв и растения
Основываясь на многочисленных экспериментальных данных, показывающих зависимость распространения
Кислотность почв и растения
Основываясь на многочисленных экспериментальных данных, показывающих зависимость распространения

Кислотность почв и растения
Жозиас Браун-Бланке ‒ один из крупнейших геоботаников XX века, основатель
Кислотность почв и растения
Жозиас Браун-Бланке ‒ один из крупнейших геоботаников XX века, основатель

Кислотность почв и растения
Михаил Владимирович Марков считал, что растения и растительные сообщества,
Кислотность почв и растения
Михаил Владимирович Марков считал, что растения и растительные сообщества,

Кислотность почв и растения
На основании своих наблюдений и критического анализа литературных
Кислотность почв и растения
На основании своих наблюдений и критического анализа литературных

Теории кислот и оснований
Существует 3 основных теории кислот и оснований
Теории кислот и оснований
Существует 3 основных теории кислот и оснований

Теория Аррениуса-Оствальда
Сванте Август Аррениус – шведский физико-химик, автор теории электролитической
Теория Аррениуса-Оствальда
Сванте Август Аррениус – шведский физико-химик, автор теории электролитической

Теория Аррениуса-Оствальда
Согласно теории Сванте Августа Аррениуса и Вильгельма Фридриха Оствальда
Кислоты
Теория Аррениуса-Оствальда
Согласно теории Сванте Августа Аррениуса и Вильгельма Фридриха Оствальда
Кислоты

Теория Брёнстеда-Лоури
Йоханнес-Николаус Брёнстед – датский физикохимик, и Томас Мартин Лаури
Теория Брёнстеда-Лоури
Йоханнес-Николаус Брёнстед – датский физикохимик, и Томас Мартин Лаури

Теория Брёнстеда-Лоури
Согласно протонной теории (теории сопряженных кислот-оснований),
кислота – соединение, способное
Теория Брёнстеда-Лоури
Согласно протонной теории (теории сопряженных кислот-оснований),
кислота – соединение, способное

Теория Брёнстеда-Лоури
При взаимодействии кислоты и основания, кислота отдаёт свои протоны,
Теория Брёнстеда-Лоури
При взаимодействии кислоты и основания, кислота отдаёт свои протоны,

Кислотность почв
рН играет существенную роль в реакциях перехода протонов (в реакциях
Кислотность почв
рН играет существенную роль в реакциях перехода протонов (в реакциях

Кислотность почв
БРЁНСТЕДОВЫ КИСЛОТЫ − нейтральные молекулы или ионы, способные предоставлять протон
Кислотность почв
БРЁНСТЕДОВЫ КИСЛОТЫ − нейтральные молекулы или ионы, способные предоставлять протон

Теория кислот и оснований Льюиса
Гилберт Ньютон Льюис – американский физикохимик,
Теория кислот и оснований Льюиса
Гилберт Ньютон Льюис – американский физикохимик,

Теория кислот и оснований Льюиса
В теории Гилберта Ньютона Льюиса (Электронной
Теория кислот и оснований Льюиса
В теории Гилберта Ньютона Льюиса (Электронной

Теория кислот и оснований Льюиса
Кислоты по Льюису:
1) любые химические соединения,
Теория кислот и оснований Льюиса
Кислоты по Льюису:
1) любые химические соединения,

Теория кислот и оснований Льюиса
Электронные орбитали могут быть молекулярными орбитами,
Теория кислот и оснований Льюиса
Электронные орбитали могут быть молекулярными орбитами,

Теория кислот и оснований Льюиса
ЛЬЮИСОВЫ КИСЛОТЫ ЖЁСТКИЕ (СИЛЬНЫЕ) ‒
1) молекулярная
Теория кислот и оснований Льюиса
ЛЬЮИСОВЫ КИСЛОТЫ ЖЁСТКИЕ (СИЛЬНЫЕ) ‒
1) молекулярная

Теория кислот и оснований Льюиса
Внутри группы жёстких (сильных) льюисовых кислот
Теория кислот и оснований Льюиса
Внутри группы жёстких (сильных) льюисовых кислот

Теория кислот и оснований Льюиса
Оценить силу льюисовских кислот можно по
Теория кислот и оснований Льюиса
Оценить силу льюисовских кислот можно по

Теория кислот и оснований Льюиса
ЛЬЮИСОВЫ КИСЛОТЫ МЯГКИЕ (СЛАБЫЕ) ‒
1) частицы
Теория кислот и оснований Льюиса
ЛЬЮИСОВЫ КИСЛОТЫ МЯГКИЕ (СЛАБЫЕ) ‒
1) частицы

Теория кислот и оснований Льюиса
Примерами мягких (слабых) кислот по Льюису
Теория кислот и оснований Льюиса
Примерами мягких (слабых) кислот по Льюису

Теория кислот и оснований Льюиса
Список сильных и слабых кислот по
Теория кислот и оснований Льюиса
Список сильных и слабых кислот по

Теория кислот и оснований Льюиса
ЛЬЮИСОВЫ ОСНОВАНИЯ ‒
1) соединения, возбуждающие реакцию
Теория кислот и оснований Льюиса
ЛЬЮИСОВЫ ОСНОВАНИЯ ‒
1) соединения, возбуждающие реакцию

Теория кислот и оснований Льюиса
К льюисовым основаниям относятся Н2О, оксианионы
Теория кислот и оснований Льюиса
К льюисовым основаниям относятся Н2О, оксианионы

Теория кислот и оснований Льюиса
ЛЬЮИСОВЫ ОСНОВАНИЯ ЖЁСТКИЕ (СИЛЬНЫЕ) −
1) сильно
Теория кислот и оснований Льюиса
ЛЬЮИСОВЫ ОСНОВАНИЯ ЖЁСТКИЕ (СИЛЬНЫЕ) −
1) сильно

Теория кислот и оснований Льюиса
В жёстких (сильных) льюисовых основаниях электронные
Теория кислот и оснований Льюиса
В жёстких (сильных) льюисовых основаниях электронные

Теория кислот и оснований Льюиса
ЛЬЮИСОВЫ ОСНОВАНИЯ МЯГКИЕ (СЛАБЫЕ) ‒ частицы
Теория кислот и оснований Льюиса
ЛЬЮИСОВЫ ОСНОВАНИЯ МЯГКИЕ (СЛАБЫЕ) ‒ частицы

Теория кислот и оснований Льюиса
Список сильных и слабых оснований по
Теория кислот и оснований Льюиса
Список сильных и слабых оснований по
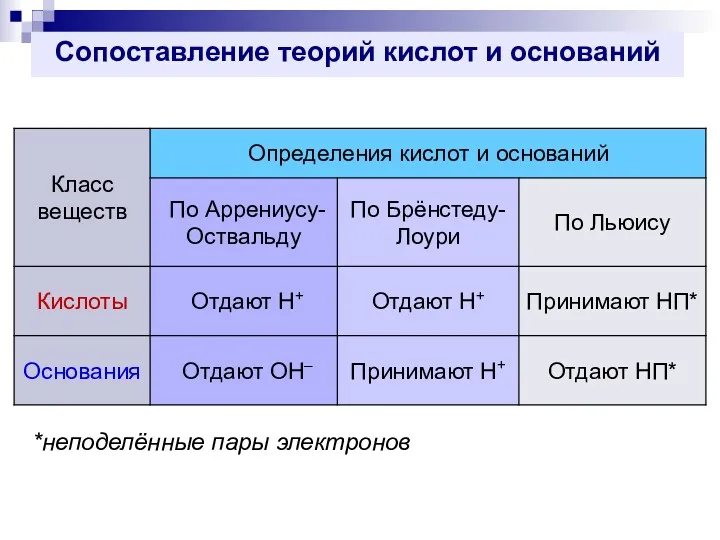
Сопоставление теорий кислот и оснований
*неподелённые пары электронов
Сопоставление теорий кислот и оснований
*неподелённые пары электронов

Буферность почв
Буферность почвы − способность почвы противостоять изменению её свойств при
Буферность почв
Буферность почвы − способность почвы противостоять изменению её свойств при

Буферность почв
Буферность почвы кислотно-щелочная – способность жидкой и твёрдой фаз почвы
Буферность почв
Буферность почвы кислотно-щелочная – способность жидкой и твёрдой фаз почвы

Буферность почв
Высокая буферность проявляется в тех случаях, когда концентрации компонентов буферного
Буферность почв
Высокая буферность проявляется в тех случаях, когда концентрации компонентов буферного

Буферность почв
Уравнение Гендерсона–Хассельбаха – логарифмическая форма уравнения константы равновесия диссоциации слабых
Буферность почв
Уравнение Гендерсона–Хассельбаха – логарифмическая форма уравнения константы равновесия диссоциации слабых

Буферность почв
Аналогичное уравнение может быть выведено и для щелочных растворов:
,
где
Буферность почв
Аналогичное уравнение может быть выведено и для щелочных растворов:
,
где

Буферность почв
Буферная ёмкость – количество сильной кислоты (или щелочи), которое надо
Буферность почв
Буферная ёмкость – количество сильной кислоты (или щелочи), которое надо

Буферность почв
Буферные кислотно-щелочные системы – системы способные противостоять изменению рН при
Буферность почв
Буферные кислотно-щелочные системы – системы способные противостоять изменению рН при

Буферность почв
Свойства буферных кислотно-щелочных систем, содержащих слабую кислоту (или слабое основание)
Буферность почв
Свойства буферных кислотно-щелочных систем, содержащих слабую кислоту (или слабое основание)

Буферность почв
Если буферная кислотно-щелочная система представлена слабым основанием и его солью,
Буферность почв
Если буферная кислотно-щелочная система представлена слабым основанием и его солью,

Буферность почв
Примеры буферных кислотно-щелочных систем:
а) ацетатной буферной смесь: СН3СООН – Н2О
Буферность почв
Примеры буферных кислотно-щелочных систем:
а) ацетатной буферной смесь: СН3СООН – Н2О

Буферность почв
Буферные кислотно-щелочные системы, как известно, обладают способностью поддерживать рН на
Буферность почв
Буферные кислотно-щелочные системы, как известно, обладают способностью поддерживать рН на

Механизм кислотно-основной буферности почв (по Ульриху)
Механизм кислотно-основной буферности почв (по Ульриху)

Буферность почв
Изотоническая буферность почв − буферность, позволяющая поддерживать постоянное осмотическое давление
Буферность почв
Изотоническая буферность почв − буферность, позволяющая поддерживать постоянное осмотическое давление

Буферность почв
Окислительно-восстановительная буферность почв − буферность, позволяющая поддерживать окислительно-восстановительный потенциал.
Окислительно-восстановительный потенциал
Буферность почв
Окислительно-восстановительная буферность почв − буферность, позволяющая поддерживать окислительно-восстановительный потенциал.
Окислительно-восстановительный потенциал
 Типы биотехнологических процессов. Биореакторы. Отделение, очистка и модификация продуктов
Типы биотехнологических процессов. Биореакторы. Отделение, очистка и модификация продуктов Доказательства происхождения животных
Доказательства происхождения животных Рыбы. Систематика рыб
Рыбы. Систематика рыб методическая разработка урока биологии в 6 классе по теме Плоды
методическая разработка урока биологии в 6 классе по теме Плоды Растение бегония
Растение бегония Різноманітність клітин людського організму. Тканини
Різноманітність клітин людського організму. Тканини Класс Пресмыкающиеся. Класс Птицы. Класс Млекопитающие
Класс Пресмыкающиеся. Класс Птицы. Класс Млекопитающие Развитие производных эктодермы
Развитие производных эктодермы Семейство пингвины
Семейство пингвины История открытия микроорганизмов
История открытия микроорганизмов Грибы. Окружающий мир 3 класс (по учебнику Н.Ф.Виноградовой, Г.С. Калиновой)
Грибы. Окружающий мир 3 класс (по учебнику Н.Ф.Виноградовой, Г.С. Калиновой) Биология как наука
Биология как наука Отдел Моховидные
Отдел Моховидные Оптическая часть зрительной системы
Оптическая часть зрительной системы Антропогенез. Основные источники информации. Человек и обезьяны. Эволюция приматов
Антропогенез. Основные источники информации. Человек и обезьяны. Эволюция приматов Ищите доктора в природе
Ищите доктора в природе Анализаторы
Анализаторы Антропогенез. Расы. Расизм. Часть 2
Антропогенез. Расы. Расизм. Часть 2 Пищеварительный процесс
Пищеварительный процесс 10 интересных фактов о животных
10 интересных фактов о животных Различия в строении клеток эукариот и прокариот. Строение клеток прокариот
Различия в строении клеток эукариот и прокариот. Строение клеток прокариот Адаптация к действию высоких температур
Адаптация к действию высоких температур Животные холодных стран
Животные холодных стран Что делают животные
Что делают животные Хвойные деревья. Кустарники. Лиственные деревья
Хвойные деревья. Кустарники. Лиственные деревья Разработка и обоснование системы удобрения в севооборотах ООО Журавушка
Разработка и обоснование системы удобрения в севооборотах ООО Журавушка Растительный мир Красноярского края
Растительный мир Красноярского края Нутрициология
Нутрициология