- Обмен белков: Индивидуальные пути обмена аминокислот

Содержание
- 2. Индивидуальные пути обмена отдельных аминокислот
- 3. ОБМЕН СЕРИНА и ГЛИЦИНА
- 4. СЕРИН - заменимая АМК, может синтезироваться из промежуточного продукта гликолиза - 3-фосфоглицерата, аминогруппу получает от глутаминовой
- 5. Основной путь катаболизма ГЛИЦИНА - обратимая реакция (связанная с использованием ТГФК), катализируется глицинсинтазой - ферментным комплексом
- 6. Пути метаболизма глицина и серина Биологическая роль глицина и серина Н4-фолат = ТГФК
- 7. Гиперглицинемия (дефект глицинрасщепляющей системы) - повреждение мозга, судороги, гипотония, нарушения дыхания. Глицинурия (до 1 г/сут, в
- 8. ОБМЕН СЕРОСОДЕРЖАЩИХ АМИНОКИСЛОТ В состав белков человека входят 2 аминокислоты, содержащие серу, - метионин и цистеин.
- 9. МЕТИОНИН - незаменимая АМК. Необходима для синтеза белков, участвует в реакциях дезаминирования, является источником серы для
- 10. S-аденозилметионин (SAM) - сульфониевая форма метионина, образующаяся при его присоединении к молекуле аденозина (продукта гидролиза АТФ)
- 11. 1. Синтез фосфатидилхолина из фосфатидил-этаноламина Фосфатидилхолины (лецитины) - наиболее распространенная группа глицерофосфолипидов, участвующих в образовании мембран
- 12. 2. Синтез карнитина - переносчика жирных кислот через мембрану митохондрий
- 13. 3. Синтез креатина необходимого для образования в мышцах высокоэнергетического соединения - креатинфосфата. Синтез креатина идет в
- 14. Креатин с кровотоком переносится в мышцы и в клетки головного мозга, где из него образуется высокоэнергетическое
- 15. Креатинкиназа локализована в цитозоле и в митохондриях клеток, обладает органоспецифичностью Известны 3 изоформы креатинкиназы: ВВ -
- 16. РЕАКЦИИ ТРАНСМЕТИЛИРОВАНИЯ используются также для: Синтеза адреналина из норадреналина Синтеза анзерина из карнозина Метилирования азотистых оснований
- 17. РЕГЕНЕРАЦИЯ МЕТИОНИНА В результате отщепления метильной группы SAM превращается в S-аденозилгомоцистеин (SAГ), который при действии гидроксилазы
- 18. МЕТИОНИН - незаменимая АМК, однако она может регенерироваться из гомоцистеина. Следовательно, незаменим именно гомоцистеин, но единственным
- 19. Функции цистеина - участие в фолдинге белков за счет способности тиогруппы цистеина образовывать дисульфидные связи. При
- 20. СИНТЕЗ ТАУРИНА- важный путь использования цистеина, который осуществляется за счет декарбоксилирования производных цистеина - цистеиновой и
- 21. Глутатион
- 22. ОБМЕН ФЕНИЛАЛАНИНА И ТИРОЗИНА
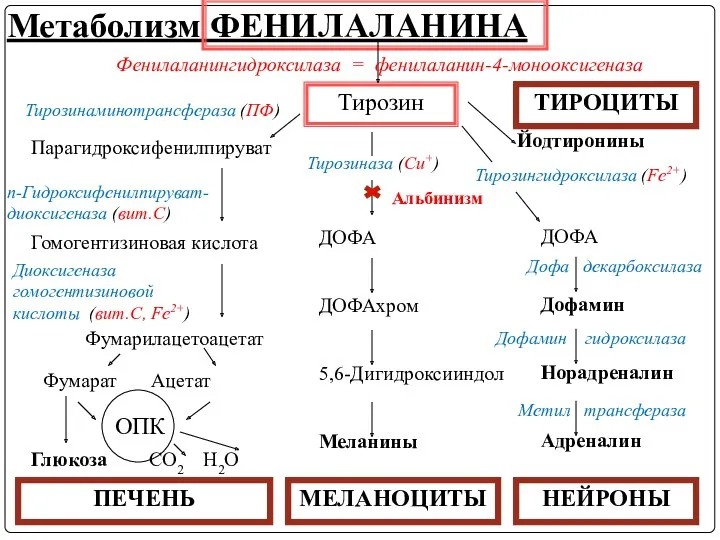
- 23. Метаболизм ФЕНИЛАЛАНИНА Фенилаланин - незаменимая АМК 2 основных пути метаболизма: включение в белки и превращение в
- 24. Метаболизм ФЕНИЛАЛАНИНА Тирозин Фенилаланингидроксилаза = фенилаланин-4-монооксигеназа Тирозинаминотрансфераза (ПФ) Парагидроксифенилпируват Гомогентизиновая кислота п-Гидроксифенилпируват-диоксигеназа (вит.С) Диоксигеназа гомогентизиновой кислоты
- 25. Обмен ФЕНИЛАЛАНИНА и ТИРОЗИНА связан со значительным количеством реакций гидроксилирования, катализируемых оксигеназами (гидроксилазами), использующими молекулу О2
- 26. Реакции гидроксилирования фенилаланина (1) и регенерации Н4БП (2)
- 27. Дефект фенилаланингидроксилазы приводит к развитию наследственного заболевания фенилпировиноградной олигофрении (фенилкетонурии, ФКУ) Выделяют 2 формы ФКУ: классическая
- 28. Вариантная ФКУ(коферментзависимая гиперфенилаланинемия, «злокачественная» ФКУ): Причина - мутации генов, контролирующих синтез Н4БП. Частота заболевания 1- 2
- 29. Пути катаболизма фенилаланина при дефекте фенилаланин-4-монооксигеназы
- 30. Высокие концентрации фенилаланина ограничивают транспорт Тир и Трп через гематоэнцефалический барьер и тормозят синтез нейромедиаторов (дофамина,
- 31. Диагностика у гетерозиготных родителей: детекция наличия дефектного гена с помощью: а. теста на толерантность к Фен
- 32. Метаболизм тирозина обладает органоспецифичностью Тирозин в разных тканях выступает предшественником таких соединений, как катехоламины, тироксин, меланины
- 33. Метаболизм ФЕНИЛАЛАНИНА Тирозин Тирозинаминотрансфераза (ПФ) Парагидроксифенилпируват Гомогентизиновая кислота п-Гидроксифенилпируват-диоксигеназа (вит.С) Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+) Фумарилацетоацетат
- 34. Катаболизм тирозина в печени В печени происходит катаболизм тирозина до конечных продуктов. Специфический путь катаболизма включает
- 35. ПАТОЛОГИИ, СВЯЗАННЫЕ С НАРУШЕНИЕМ МЕТАБОЛИЗМА ТИРОЗИНА В ПЕЧЕНИ
- 36. Метаболизм ФЕНИЛАЛАНИНА Тирозин Тирозинаминотрансфераза (ПФ) Парагидроксифенилпируват Гомогентизиновая кислота п-Гидроксифенилпируват-диоксигеназа (вит.С) Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+) Фумарилацетоацетат
- 37. В ходе метаболических превращений Тир могут наблюдаться ряд нарушений, имеющих наследственный характер: 1. Алкаптонурия («черная моча»)
- 38. 2. Тирозиноз (тирозинемия типа I) Причина заболевания - дефект гена фумарилацетоацетат-гидролазы, катализирующей расщепление фумарилацетоацетата на фумарат
- 39. 3. Синдром Рихнера-Ханхорта (тирозинемия типа II) Причина – дефект фермента тирозинаминотрансферазы, катализирующей образование n-оксифенилпировиноградной кислоты из
- 40. 4. Тирозинемия новорожденных (кратковременная) Возникает в результате снижения активности фермента n-гидроксифенилпируватдиоксигеназы, превращающего n-гидроксифенилпируват в гомогентизиновую кислоту
- 41. ПРЕВРАЩЕНИЕ ТИРОЗИНА В МЕЛАНОЦИТАХ
- 42. В пигментных клетках (меланоцитах) тирозин выступает предшественником тёмных пигментов - меланинов. Среди них преобладают 2 типа:
- 43. СХЕМА МЕТАБОЛИЗМА ТИРОЗИНА В МЕЛАНОЦИТАХ
- 44. Метаболизм ФЕНИЛАЛАНИНА Тирозин Тирозинаминотрансфераза (ПФ) Парагидроксифенилпируват Гомогентизиновая кислота п-Гидроксифенилпируват-диоксигеназа (вит.С) Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+) Фумарилацетоацетат
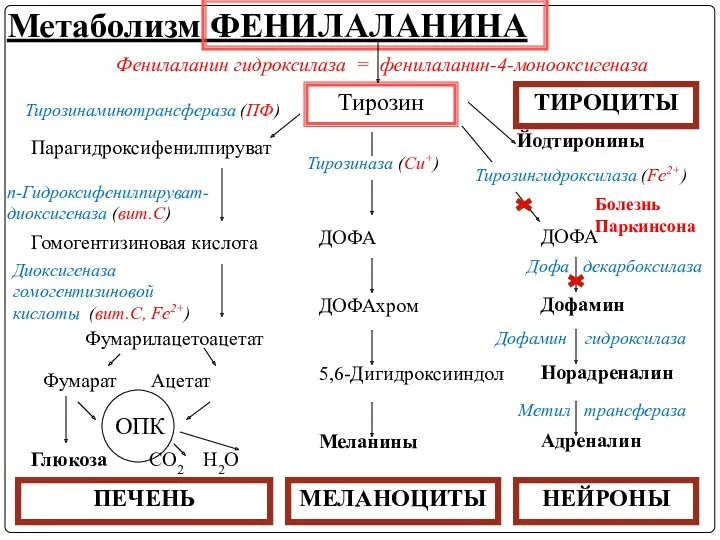
- 45. Альбинизм При альбинизме глаз и кожи, негативном по тирозиназе наблюдается – врожденный дефект гена тирозиназы, катализирующей
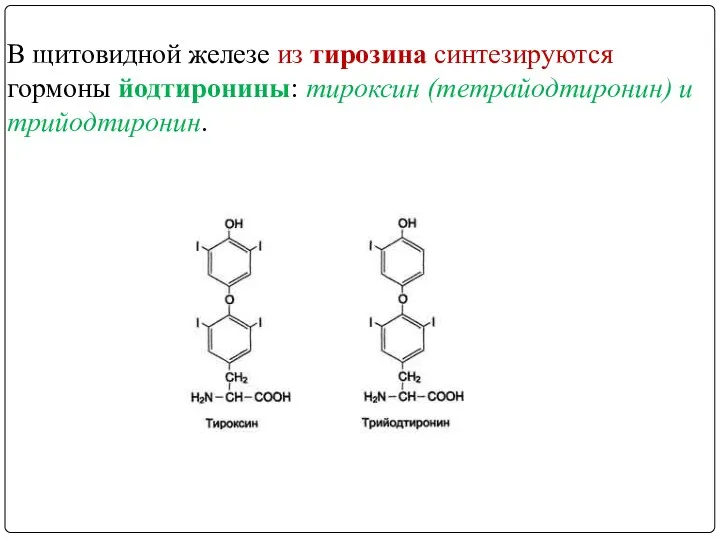
- 46. МЕТАБОЛИЗМ ТИРОЗИНА В ЩИТОВИДНОЙ ЖЕЛЕЗЕ
- 47. В щитовидной железе из тирозина синтезируются гормоны йодтиронины: тироксин (тетрайодтиронин) и трийодтиронин.
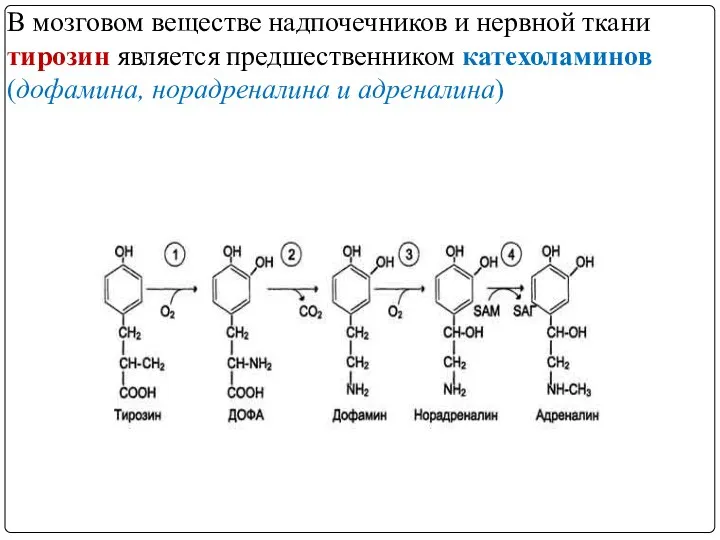
- 48. МЕТАБОЛИЗМ ТИРОЗИНА В НАДПОЧЕЧНИКАХ И НЕРВНОЙ ТКАНИ
- 49. В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов (дофамина, норадреналина и адреналина)
- 50. Тирозингидроксилаза (1) в надпочечниках и катехоламинергичес-ких нейронах - Fе2+-зависимый фермент, в качестве кофермента использующий Н4БП, катализирует
- 51. Метаболизм ФЕНИЛАЛАНИНА Тирозин Тирозинаминотрансфераза (ПФ) Парагидроксифенилпируват Гомогентизиновая кислота п-Гидроксифенилпируват-диоксигеназа (вит.С) Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+) Фумарилацетоацетат
- 52. Нарушения метаболизма тирозина вносят вклад в развитие болезни Паркинсона, развивающейся при недостаточности дофамина в черной субстанции
- 54. Скачать презентацию
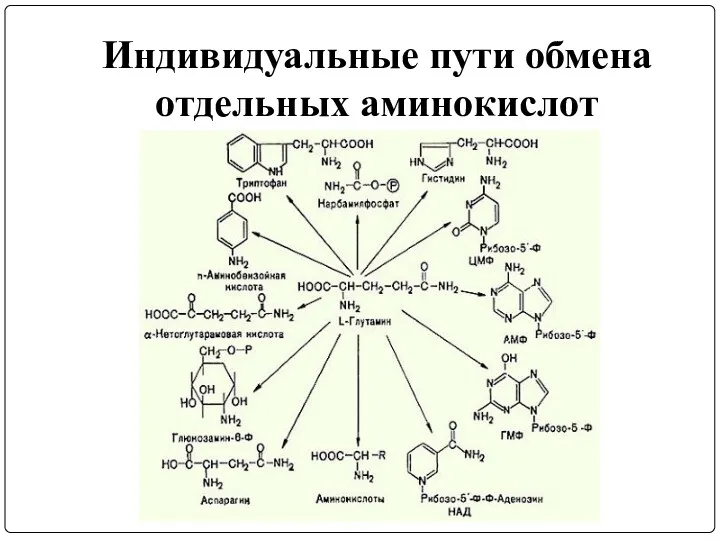
Индивидуальные пути обмена
отдельных аминокислот
Индивидуальные пути обмена
отдельных аминокислот

ОБМЕН СЕРИНА и ГЛИЦИНА
ОБМЕН СЕРИНА и ГЛИЦИНА
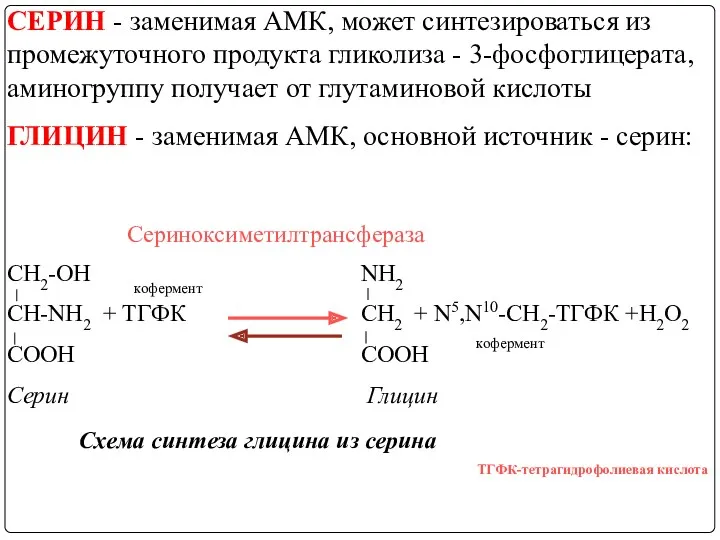
СЕРИН - заменимая АМК, может синтезироваться из промежуточного продукта гликолиза -
СЕРИН - заменимая АМК, может синтезироваться из промежуточного продукта гликолиза -

Основной путь катаболизма ГЛИЦИНА - обратимая реакция (связанная с использованием ТГФК),
Основной путь катаболизма ГЛИЦИНА - обратимая реакция (связанная с использованием ТГФК),
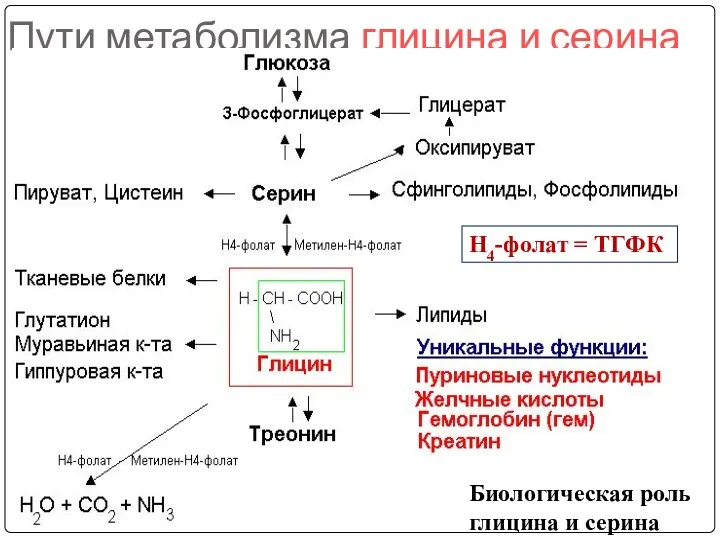
Пути метаболизма глицина и серина
Биологическая роль глицина и серина
Н4-фолат = ТГФК
Пути метаболизма глицина и серина
Биологическая роль глицина и серина
Н4-фолат = ТГФК

Гиперглицинемия (дефект глицинрасщепляющей системы) - повреждение мозга, судороги, гипотония, нарушения дыхания.
Глицинурия
Гиперглицинемия (дефект глицинрасщепляющей системы) - повреждение мозга, судороги, гипотония, нарушения дыхания.
Глицинурия

ОБМЕН СЕРОСОДЕРЖАЩИХ АМИНОКИСЛОТ
В состав белков человека входят 2 аминокислоты, содержащие серу,
ОБМЕН СЕРОСОДЕРЖАЩИХ АМИНОКИСЛОТ
В состав белков человека входят 2 аминокислоты, содержащие серу,

МЕТИОНИН - незаменимая АМК. Необходима для синтеза белков, участвует в реакциях
МЕТИОНИН - незаменимая АМК. Необходима для синтеза белков, участвует в реакциях
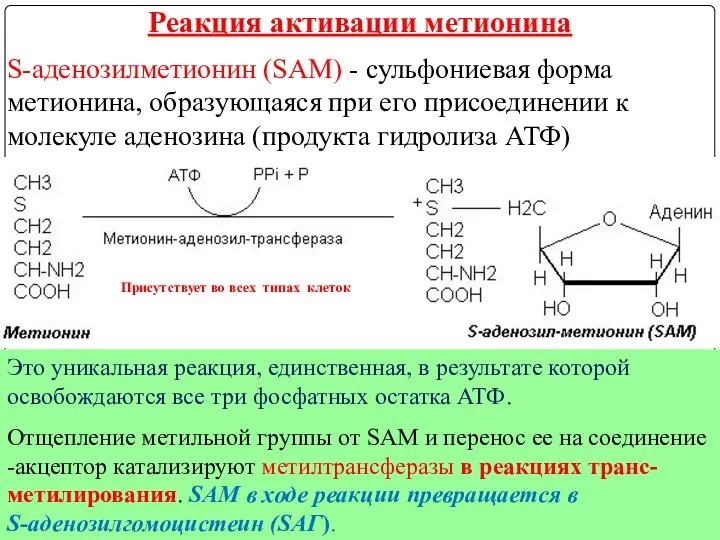
S-аденозилметионин (SAM) - сульфониевая форма метионина, образующаяся при его присоединении к
S-аденозилметионин (SAM) - сульфониевая форма метионина, образующаяся при его присоединении к
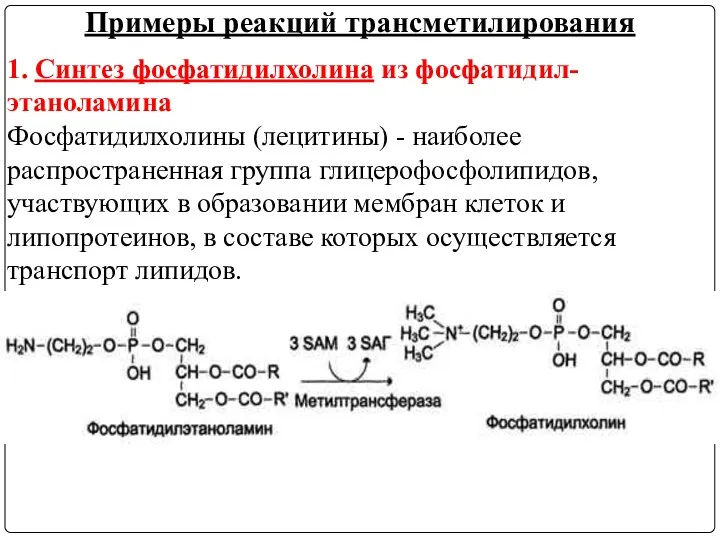
1. Синтез фосфатидилхолина из фосфатидил-этаноламина
Фосфатидилхолины (лецитины) - наиболее распространенная группа глицерофосфолипидов,
1. Синтез фосфатидилхолина из фосфатидил-этаноламина
Фосфатидилхолины (лецитины) - наиболее распространенная группа глицерофосфолипидов,
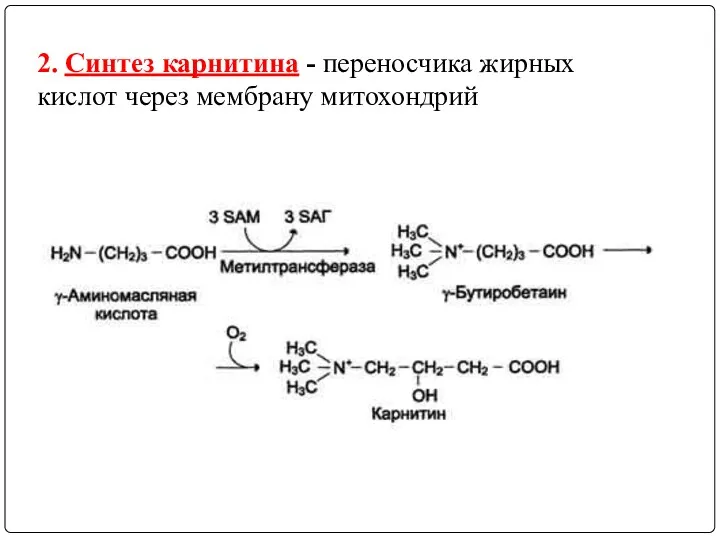
2. Синтез карнитина - переносчика жирных кислот через мембрану митохондрий
2. Синтез карнитина - переносчика жирных кислот через мембрану митохондрий

3. Синтез креатина необходимого для образования в мышцах высокоэнергетического соединения -
3. Синтез креатина необходимого для образования в мышцах высокоэнергетического соединения -
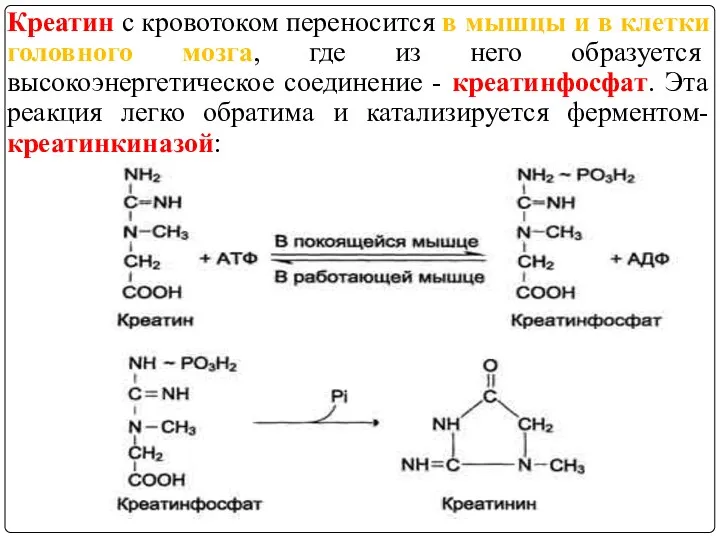
Креатин с кровотоком переносится в мышцы и в клетки головного мозга,
Креатин с кровотоком переносится в мышцы и в клетки головного мозга,

Креатинкиназа локализована в цитозоле и в митохондриях клеток, обладает органоспецифичностью
Известны 3
Креатинкиназа локализована в цитозоле и в митохондриях клеток, обладает органоспецифичностью
Известны 3

РЕАКЦИИ ТРАНСМЕТИЛИРОВАНИЯ
используются также для:
Синтеза адреналина из норадреналина
Синтеза анзерина
РЕАКЦИИ ТРАНСМЕТИЛИРОВАНИЯ
используются также для:
Синтеза адреналина из норадреналина
Синтеза анзерина

РЕГЕНЕРАЦИЯ МЕТИОНИНА
В результате отщепления метильной группы SAM превращается в S-аденозилгомоцистеин (SAГ),
РЕГЕНЕРАЦИЯ МЕТИОНИНА
В результате отщепления метильной группы SAM превращается в S-аденозилгомоцистеин (SAГ),

МЕТИОНИН - незаменимая АМК, однако она может регенерироваться из гомоцистеина.
Следовательно,
МЕТИОНИН - незаменимая АМК, однако она может регенерироваться из гомоцистеина.
Следовательно,
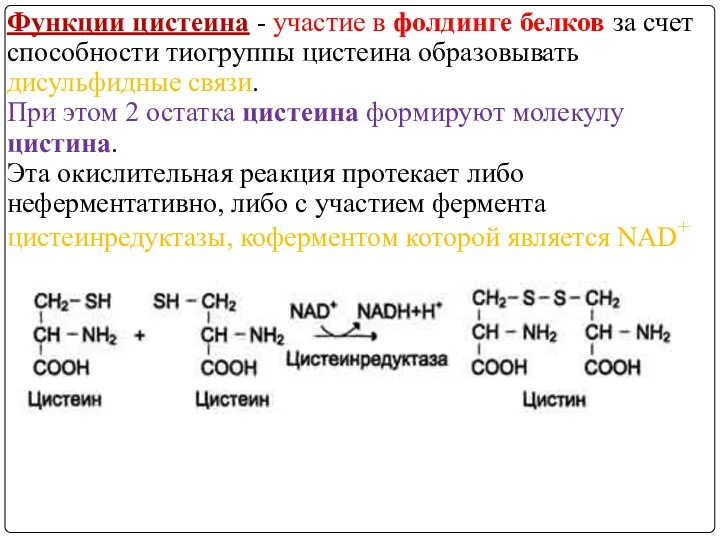
Функции цистеина - участие в фолдинге белков за счет способности тиогруппы
Функции цистеина - участие в фолдинге белков за счет способности тиогруппы
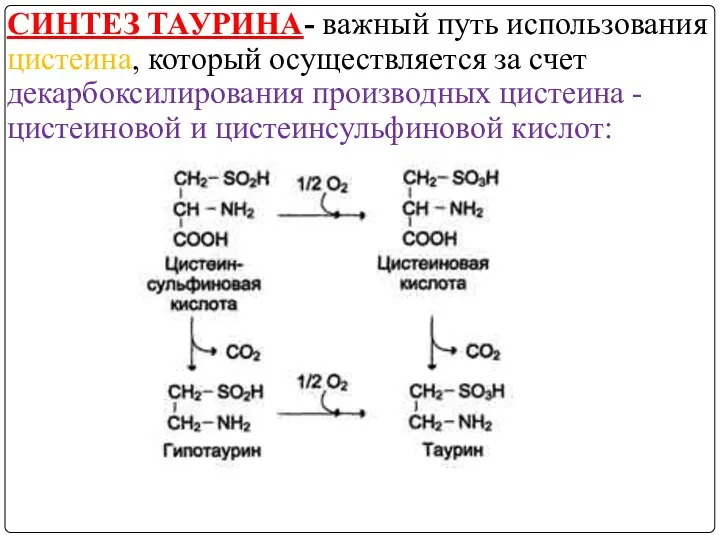
СИНТЕЗ ТАУРИНА- важный путь использования цистеина, который осуществляется за счет декарбоксилирования
СИНТЕЗ ТАУРИНА- важный путь использования цистеина, который осуществляется за счет декарбоксилирования
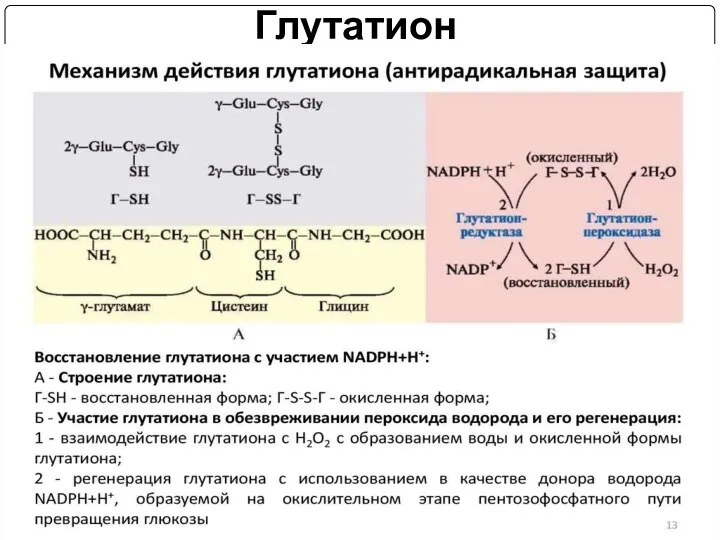
Глутатион
Глутатион

ОБМЕН ФЕНИЛАЛАНИНА И ТИРОЗИНА
ОБМЕН ФЕНИЛАЛАНИНА И ТИРОЗИНА

Метаболизм ФЕНИЛАЛАНИНА
Фенилаланин - незаменимая АМК
2 основных пути метаболизма: включение в
Метаболизм ФЕНИЛАЛАНИНА
Фенилаланин - незаменимая АМК
2 основных пути метаболизма: включение в
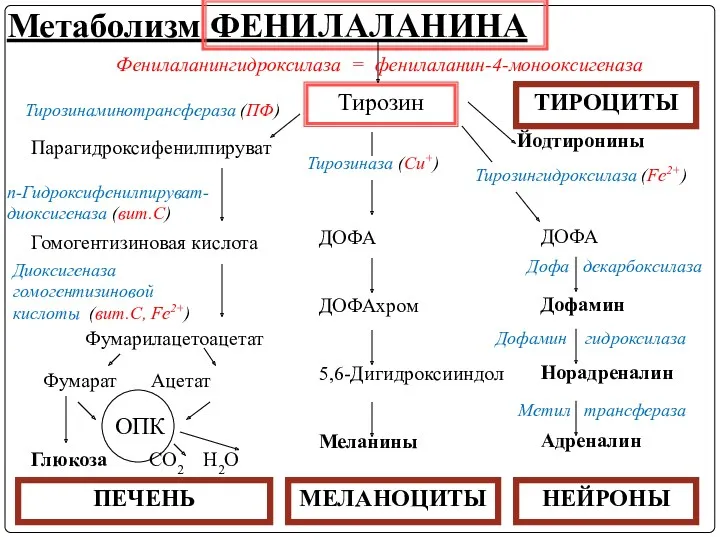
Метаболизм ФЕНИЛАЛАНИНА
Тирозин
Фенилаланингидроксилаза = фенилаланин-4-монооксигеназа
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
Гомогентизиновая кислота
п-Гидроксифенилпируват-диоксигеназа (вит.С)
Диоксигеназа гомогентизиновой кислоты
Метаболизм ФЕНИЛАЛАНИНА
Тирозин
Фенилаланингидроксилаза = фенилаланин-4-монооксигеназа
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
Гомогентизиновая кислота
п-Гидроксифенилпируват-диоксигеназа (вит.С)
Диоксигеназа гомогентизиновой кислоты

Обмен ФЕНИЛАЛАНИНА и ТИРОЗИНА связан со значительным количеством реакций гидроксилирования, катализируемых
Обмен ФЕНИЛАЛАНИНА и ТИРОЗИНА связан со значительным количеством реакций гидроксилирования, катализируемых
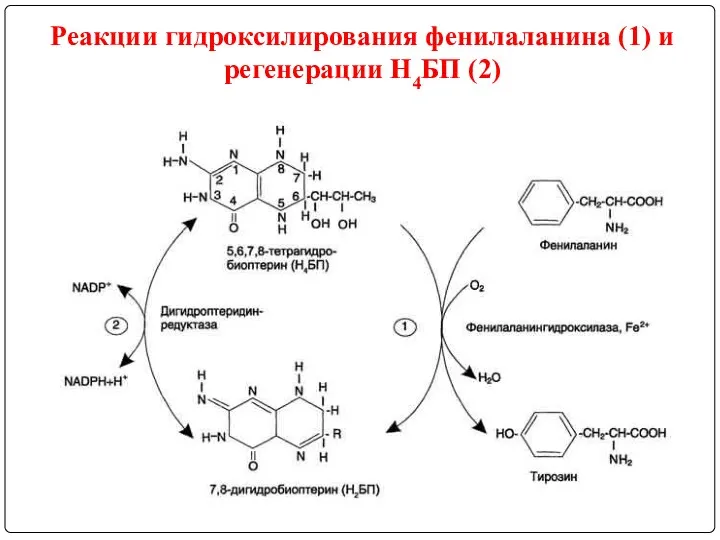
Реакции гидроксилирования фенилаланина (1) и регенерации Н4БП (2)
Реакции гидроксилирования фенилаланина (1) и регенерации Н4БП (2)

Дефект фенилаланингидроксилазы приводит к развитию наследственного заболевания фенилпировиноградной олигофрении (фенилкетонурии, ФКУ)
Дефект фенилаланингидроксилазы приводит к развитию наследственного заболевания фенилпировиноградной олигофрении (фенилкетонурии, ФКУ)

Вариантная ФКУ(коферментзависимая гиперфенилаланинемия, «злокачественная» ФКУ):
Причина - мутации генов, контролирующих синтез Н4БП.
Вариантная ФКУ(коферментзависимая гиперфенилаланинемия, «злокачественная» ФКУ):
Причина - мутации генов, контролирующих синтез Н4БП.
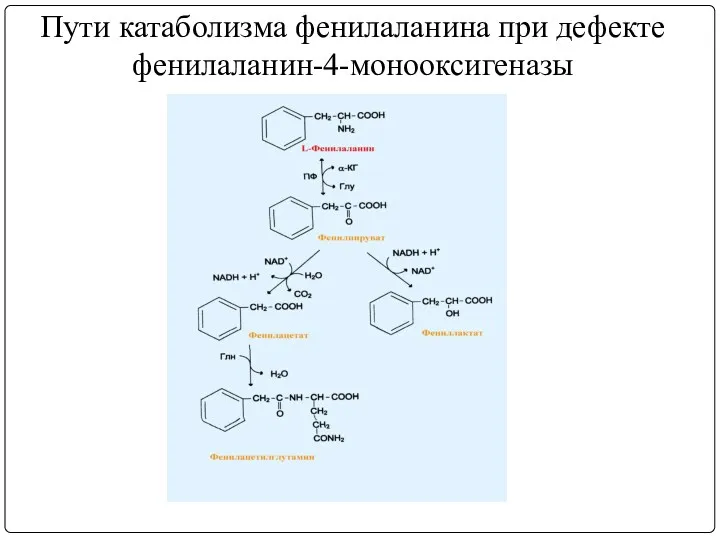
Пути катаболизма фенилаланина при дефекте
фенилаланин-4-монооксигеназы
Пути катаболизма фенилаланина при дефекте
фенилаланин-4-монооксигеназы

Высокие концентрации фенилаланина ограничивают транспорт Тир и Трп через гематоэнцефалический барьер
Высокие концентрации фенилаланина ограничивают транспорт Тир и Трп через гематоэнцефалический барьер

Диагностика
у гетерозиготных родителей:
детекция наличия дефектного гена с помощью:
а. теста
Диагностика
у гетерозиготных родителей:
детекция наличия дефектного гена с помощью:
а. теста

Метаболизм тирозина обладает органоспецифичностью
Тирозин в разных тканях выступает предшественником таких соединений,
Метаболизм тирозина обладает органоспецифичностью
Тирозин в разных тканях выступает предшественником таких соединений,
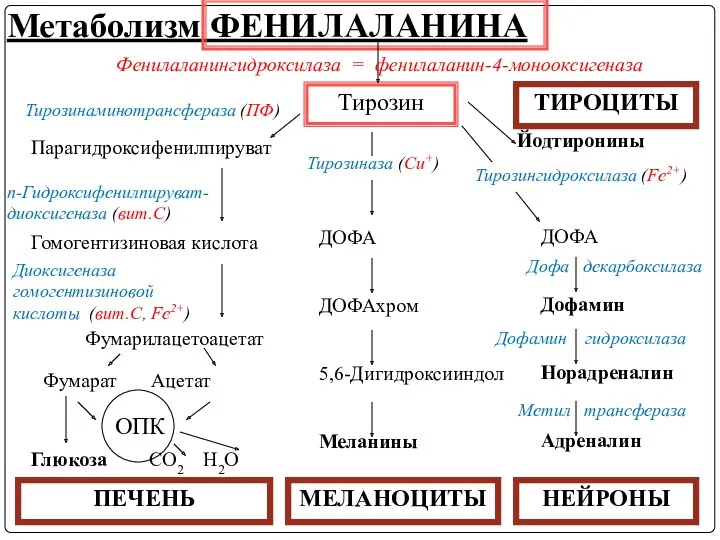
Метаболизм ФЕНИЛАЛАНИНА
Тирозин
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
Гомогентизиновая кислота
п-Гидроксифенилпируват-диоксигеназа (вит.С)
Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+)
Фумарилацетоацетат
Фумарат
Метаболизм ФЕНИЛАЛАНИНА
Тирозин
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
Гомогентизиновая кислота
п-Гидроксифенилпируват-диоксигеназа (вит.С)
Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+)
Фумарилацетоацетат
Фумарат

Катаболизм тирозина в печени
В печени происходит катаболизм тирозина до конечных
Катаболизм тирозина в печени
В печени происходит катаболизм тирозина до конечных

ПАТОЛОГИИ, СВЯЗАННЫЕ С НАРУШЕНИЕМ МЕТАБОЛИЗМА ТИРОЗИНА В ПЕЧЕНИ
ПАТОЛОГИИ, СВЯЗАННЫЕ С НАРУШЕНИЕМ МЕТАБОЛИЗМА ТИРОЗИНА В ПЕЧЕНИ
Метаболизм ФЕНИЛАЛАНИНА
Тирозин
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
Гомогентизиновая кислота
п-Гидроксифенилпируват-диоксигеназа (вит.С)
Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+)
Фумарилацетоацетат
Фумарат
Метаболизм ФЕНИЛАЛАНИНА
Тирозин
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
Гомогентизиновая кислота
п-Гидроксифенилпируват-диоксигеназа (вит.С)
Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+)
Фумарилацетоацетат
Фумарат

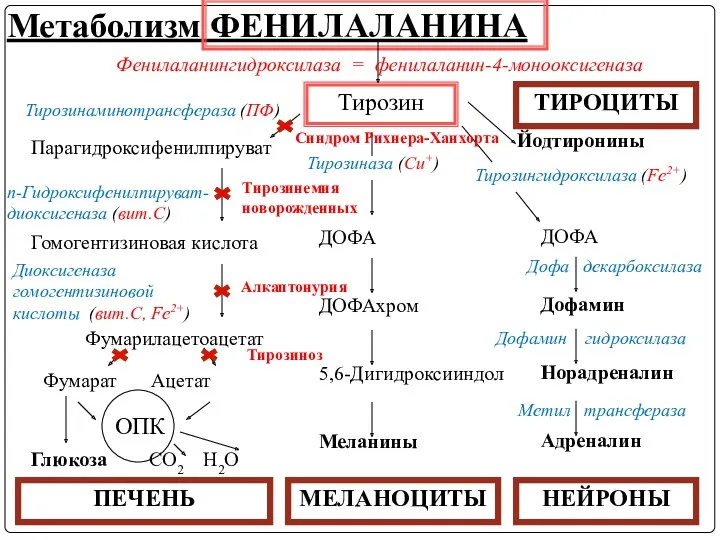
В ходе метаболических превращений Тир могут наблюдаться ряд нарушений, имеющих наследственный
В ходе метаболических превращений Тир могут наблюдаться ряд нарушений, имеющих наследственный

2. Тирозиноз (тирозинемия типа I)
Причина заболевания - дефект гена фумарилацетоацетат-гидролазы, катализирующей
2. Тирозиноз (тирозинемия типа I)
Причина заболевания - дефект гена фумарилацетоацетат-гидролазы, катализирующей

3. Синдром Рихнера-Ханхорта (тирозинемия типа II)
Причина – дефект фермента тирозинаминотрансферазы, катализирующей
3. Синдром Рихнера-Ханхорта (тирозинемия типа II)
Причина – дефект фермента тирозинаминотрансферазы, катализирующей

4. Тирозинемия новорожденных (кратковременная)
Возникает в результате снижения активности фермента n-гидроксифенилпируватдиоксигеназы, превращающего
4. Тирозинемия новорожденных (кратковременная)
Возникает в результате снижения активности фермента n-гидроксифенилпируватдиоксигеназы, превращающего

ПРЕВРАЩЕНИЕ ТИРОЗИНА В МЕЛАНОЦИТАХ
ПРЕВРАЩЕНИЕ ТИРОЗИНА В МЕЛАНОЦИТАХ

В пигментных клетках (меланоцитах) тирозин выступает предшественником тёмных пигментов - меланинов.
В пигментных клетках (меланоцитах) тирозин выступает предшественником тёмных пигментов - меланинов.
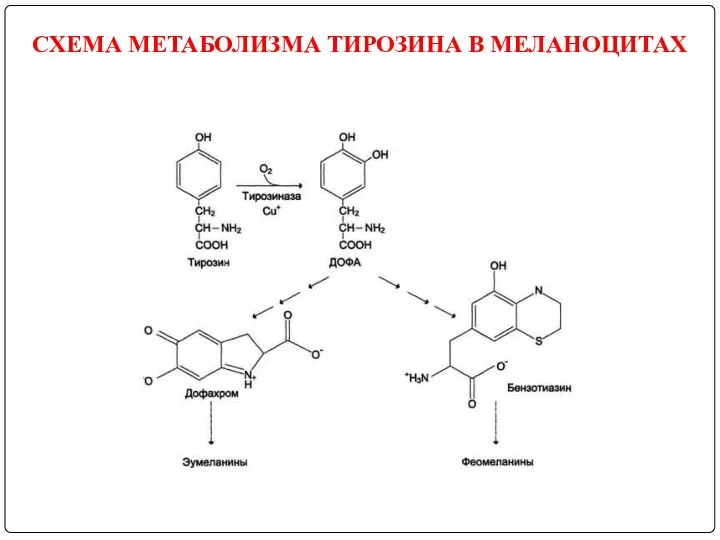
СХЕМА МЕТАБОЛИЗМА ТИРОЗИНА В МЕЛАНОЦИТАХ
СХЕМА МЕТАБОЛИЗМА ТИРОЗИНА В МЕЛАНОЦИТАХ
Метаболизм ФЕНИЛАЛАНИНА
Тирозин
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
Гомогентизиновая кислота
п-Гидроксифенилпируват-диоксигеназа (вит.С)
Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+)
Фумарилацетоацетат
Фумарат
Метаболизм ФЕНИЛАЛАНИНА
Тирозин
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
Гомогентизиновая кислота
п-Гидроксифенилпируват-диоксигеназа (вит.С)
Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+)
Фумарилацетоацетат
Фумарат

Альбинизм
При альбинизме глаз и кожи, негативном по тирозиназе наблюдается – врожденный
Альбинизм
При альбинизме глаз и кожи, негативном по тирозиназе наблюдается – врожденный

МЕТАБОЛИЗМ ТИРОЗИНА В ЩИТОВИДНОЙ ЖЕЛЕЗЕ
МЕТАБОЛИЗМ ТИРОЗИНА В ЩИТОВИДНОЙ ЖЕЛЕЗЕ
В щитовидной железе из тирозина синтезируются гормоны йодтиронины: тироксин (тетрайодтиронин) и

МЕТАБОЛИЗМ ТИРОЗИНА В НАДПОЧЕЧНИКАХ И НЕРВНОЙ ТКАНИ
МЕТАБОЛИЗМ ТИРОЗИНА В НАДПОЧЕЧНИКАХ И НЕРВНОЙ ТКАНИ
В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов
В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов

Тирозингидроксилаза (1) в надпочечниках и катехоламинергичес-ких нейронах - Fе2+-зависимый фермент, в
Тирозингидроксилаза (1) в надпочечниках и катехоламинергичес-ких нейронах - Fе2+-зависимый фермент, в
Метаболизм ФЕНИЛАЛАНИНА
Тирозин
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
Гомогентизиновая кислота
п-Гидроксифенилпируват-диоксигеназа (вит.С)
Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+)
Фумарилацетоацетат
Фумарат
Метаболизм ФЕНИЛАЛАНИНА
Тирозин
Тирозинаминотрансфераза (ПФ)
Парагидроксифенилпируват
Гомогентизиновая кислота
п-Гидроксифенилпируват-диоксигеназа (вит.С)
Диоксигеназа гомогентизиновой кислоты (вит.С, Fe2+)
Фумарилацетоацетат
Фумарат

Нарушения метаболизма тирозина вносят вклад в развитие болезни Паркинсона, развивающейся при
Нарушения метаболизма тирозина вносят вклад в развитие болезни Паркинсона, развивающейся при
 Бесполое размножение. Митоз
Бесполое размножение. Митоз Процесс выращивания салата Одесский кучерявец
Процесс выращивания салата Одесский кучерявец Инструктивные карточки по биологии для 5 класса.
Инструктивные карточки по биологии для 5 класса. Приобретенные формы поведения. (Лекция 3)
Приобретенные формы поведения. (Лекция 3) Вкусовая сенсорная система
Вкусовая сенсорная система Общая характеристика типа Моллюски
Общая характеристика типа Моллюски Дыхательная система
Дыхательная система Размножение человека. 8 класс. 1
Размножение человека. 8 класс. 1 Наследственные и врожденные заболевания. ЗППП
Наследственные и врожденные заболевания. ЗППП Птицы
Птицы Рефлексы: типы, механизм
Рефлексы: типы, механизм Отряд голенастые
Отряд голенастые Город будущего
Город будущего Выращивание огурцов
Выращивание огурцов Спинномозговые нервы
Спинномозговые нервы Мир животных
Мир животных Регуляция метаболизма
Регуляция метаболизма Органы выделения у животных в процессе эволюционного развития
Органы выделения у животных в процессе эволюционного развития Видообразование как результат микроэволюции
Видообразование как результат микроэволюции История развития генетики
История развития генетики Периодизация и продолжительность жизни животных
Периодизация и продолжительность жизни животных Мутация (3)
Мутация (3) Образование половых клеток
Образование половых клеток История развития биологии
История развития биологии Питание и пищеварение
Питание и пищеварение Трудный путь микроба
Трудный путь микроба Заболевания органов пищеварения и их профилактика.
Заболевания органов пищеварения и их профилактика. Сравнительное наблюдение за прорастанием семян, ростом растений
Сравнительное наблюдение за прорастанием семян, ростом растений