- Технология секвенирования генома и сборка генома. Лекция 8

Содержание
- 2. ДНК секвенирование Подход для определения нуклеотидной последовательности ДНК (дезоксирибонуклеиновой кислоты) 28.11.2018
- 3. 28.11.2018
- 4. 28.11.2018
- 5. Применение NGS 28.11.2018
- 6. Основные термины Геномные библиотеки - это коллекция геномной ДНК полученная от одного организма и подготовленная для
- 7. Первые методы секвенирования Maxam-Gilbert (1976-1977) Sanger (1977) 28.11.2018
- 8. 28.11.2018 Нуклеотид-специфическая деградация ДНК при обработке различными веществами
- 9. Секвенирование по Сенгеру (Золотой стандарт) 28.11.2018 Phi X 174 (ΦX174) бактериофаг был первым секвенированным ДНК геномом
- 10. Полногеномное секвенирование с использованием метода Сенгера 28.11.2018
- 11. Проект геном человека Размер генома – 3.2 Гб Длительность – 10 лет 1990 – 2000 Цена
- 12. Секвенирование по Сенгеру Плюсы: Относительно низкий уровень ошибок Удобное и дешевое секвенирование небольших фрагментов генома (16S
- 13. 28.11.2018
- 14. New Generation Sequencing 28.11.2018 Плюсы: Простая подготовка ДНК библиотек (пробоподготовка) Высокая производительность Низкая стоимость секвенирования Минусы:
- 15. Основные принципы подготовки ДНК библиотек Фрагментация ДНК Отбор размера Лигирование адаптора Амплификация библиотеки 28.11.2018
- 16. Стратегия полногеномного секвенирования использует NGS платформы 28.11.2018
- 17. Контиг (Contig) - группа перекрывающихся прочтений, представляющие участок генома. 28.11.2018 Contig is a group of overlapping
- 18. Scaffold (Скафолд) – реконструированная часть генома, полученная в результате анализа библиотек большого размера и правильного взаимного
- 19. 28.11.2018
- 20. Таргетное секвенирование 28.11.2018 Nature Methods 7, 111 - 118 (2010)
- 21. Индексирование (Баркодинг) Можно за один запуск секвенатора прочитать несколько геномов или геномных участков Индексы – короткие
- 22. Примеры индексов 28.11.2018
- 23. Платформы 28.11.2018
- 24. 28.11.2018
- 25. 454 Sequencing Technology 28.11.2018 Фрагментация ДНК Подготовка библиотеки Пришивание адапторов к молекулам ДНК с двух концов.
- 26. 28.11.2018 Один фрагмент = одна бусина (bead) Библиотека фрагментов ДНК прикрепляется к бусинам после денатурации ДНК.
- 27. 28.11.2018 Секвенирование начинается с присоединения праймера, потом присоединение комплементарного нуклеотида приводит к высвобождению пирофосфата, который взаимодействуя
- 28. 28.11.2018
- 29. Ion Torrent Подготовка библиотеки похожа на Roche 454 • фрагментация ДНК • Прикрепление адаптера • Эмульсионная
- 30. Ion Torrent полупроводниковое секвенирование 28.11.2018 Во время секвенирования, последовательно подаются нуклеотиды, при встраивании которых выделяются ионы
- 31. 28.11.2018 Ion Torrent полупроводниковое секвенирование Выделение ионов водорода приводит к изменению кислотности среды, что детектируются высокочувствительным
- 32. 28.11.2018 Ion Torrent полупроводниковое секвенирование
- 33. 28.11.2018 Ion Torrent полупроводниковое секвенирование
- 34. 28.11.2018
- 35. SOLiD 28.11.2018 Подготовка библиотеки похожа на Roche 454 • фрагментация ДНК • Прикрепление адаптера • Эмульсионная
- 36. SOLiD 28.11.2018
- 37. SOLiD 28.11.2018 Происходит последовательное взаимодействие олигонуклеотида, состоящего из специфичного динуклеотида, пяти неспецифичных нуклеотидов и флуорафора, что
- 38. SOLiD 28.11.2018 Для борьбы с неспецифичными нуклеотидами используют новые праймеры, которые короче на 1,2,3,4 нуклеотида (всего
- 39. Все описанные технологии обеспечивают односторонние прочтения ДНК 28.11.2018
- 40. 28.11.2018
- 41. Подготовка библиотеки ДНК 28.11.2018
- 42. Illumina Гибридизация ДНК-библиотек Генерация кластеров (ПЦР) Секвенирование синтезом 28.11.2018 http://www.youtube.com/watch?v=HMyCqWhwB8E
- 43. 28.11.2018 Illumina
- 44. 28.11.2018 Illumina
- 45. Pacific Biosciences single molecule real-time (SMRT) sequencing Одномолекулярное секвенирование в реальном времени Секвенировании без амплификации Очень
- 46. 28.11.2018
- 47. 28.11.2018
- 48. Сравнение платформ NGS 28.11.2018
- 49. Контроль качества данных 28.11.2018
- 50. 28.11.2018
- 51. Алгоритм контроля качества 28.11.2018 Проверка качества Определение проблемы Решение проблемы Проверка качества Последующий анализ
- 52. Зачем чистить данные? 28.11.2018 • Риды низкого качества • Контаминация (примесь ДНК другого организма) • Служебные
- 53. FASTA и FASTQ форматы 28.11.2018 FASTA FASTQ Линия начинающаяся с @ содержит идентификатор последовательности Последовательность Линия
- 54. Шкала качества Фред (Phred) 28.11.2018 Оценки качества нуклеотида Q определяются как величина, которая логарифмически зависит от
- 55. Таблица ASCII символов 28.11.2018
- 56. Разные Phred шкалы 28.11.2018
- 57. Cборка генома 28.11.2018
- 58. 28.11.2018
- 59. FastQC – инструмент для контроля качества данных На вход – исходные данные с секвенатора HTML отчет
- 60. FastQC 28.11.2018
- 61. FastQC: распределение качества по остаткам 28.11.2018 Плохое Хорошее У Illumina качество ридов обычно уменьшается к 3'
- 62. 28.11.2018 FastQC: распределение качества по ридам Плохое Хорошее Мы можем выделить группы ридов с низким и
- 63. 28.11.2018 FastQC: распределение качества по составу остатков Плохое Хорошее Мы можем определить адаптеры или сдвиг
- 64. 28.11.2018 FastQC: распределение ридов по GC составу Плохо Хорошо GC пики могут свидетельствовать о контаминации
- 65. 28.11.2018 FastQC: уровни дупликаций последовательностей Плохо Хорошо Высокий уровень дупликации свидетельствует об оверамплификации некоторых последовательностей при
- 66. 28.11.2018 FastQC: Overrepresented sequences Плохо Хорошо Перепредставленные последовательности могут показывать источник контаминации
- 67. 28.11.2018 FastQC: Качество ячеек Плохо Хорошо У Illumina можно определить проблемы с ячейками
- 68. Шаги препроцессинга Фильтрация данных по качеству Удаление ридов, качество которых ниже определенного порога; Обрезание части ридов,
- 69. У нас есть очищенные данные. Что дальше? Сборка de novo Сборка по референсному геному Выравнивание с
- 70. Сборка de novo 28.11.2018 Возьмем большое количество коротких секвенированных ридов и поместим их вместе, чтобы воссоздать
- 71. Секвенирование геномов с использованием коротких ридов 28.11.2018
- 72. План сборки 28.11.2018
- 73. Разноразмерные библиотеки ДНК 28.11.2018
- 74. 28.11.2018 http://lucigen.com/landingpage/matepair/
- 75. Сборка генома в идеальном случае 28.11.2018 Однородное покрытие ридами, нет ошибок и контаминации
- 76. Сборка генома в реальности 28.11.2018
- 77. 28.11.2018 Кафедра биоинформатики МБФ РНИМУ
- 78. Выбор правильной программы - сборщика геномов (ассемблер) На сколько большой геном? Существуют ли известные особенности этого
- 79. Сборщики геномов 28.11.2018
- 80. Оценка качества сборки генома Количество контигов Общая длинна всех контигов Длинна наибольшего контига Количество неправильно собранных
- 81. N50 Размер контига, который представляет из себя наиболее длинный контиг, такой, начиная с которого, все остальные
- 82. QUAST - QUality ASsesment Tool for Genome Assemblies 28.11.2018 http://quast.bioinf.spbau.ru/
- 83. 28.11.2018
- 84. Реальные графы де Брюйна 28.11.2018
- 85. Улучшение сборки генома 28.11.2018
- 86. Гибридная сборка 28.11.2018
- 87. Сборка на основе данных PacBio 28.11.2018
- 88. Получение финишного генома 28.11.2018
- 89. Зачем нужны финишные геномы? Функциональные геномные исследования требуют высококачественной, полной последовательности генома в качестве отправной точки
- 90. GOLD: Genomes OnLine Database 28.11.2018
- 91. Статистика GOLD 28.11.2018
- 92. Статистика GOLD 28.11.2018
- 93. Статистика GOLD 28.11.2018
- 94. Статистика GOLD 28.11.2018
- 95. NCBI Genome 28.11.2018
- 96. NCBI Genome 28.11.2018
- 97. NCBI Genome 28.11.2018
- 98. NCBI Genome 28.11.2018
- 99. NCBI SRA database 28.11.2018
- 101. Скачать презентацию
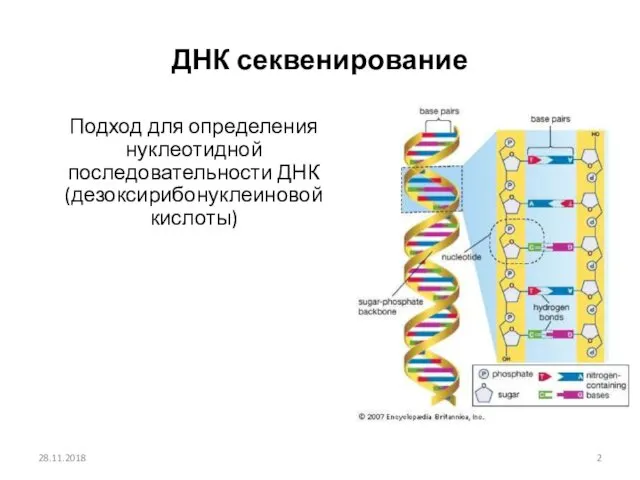
ДНК секвенирование
Подход для определения нуклеотидной последовательности ДНК (дезоксирибонуклеиновой кислоты)
28.11.2018
ДНК секвенирование
Подход для определения нуклеотидной последовательности ДНК (дезоксирибонуклеиновой кислоты)
28.11.2018
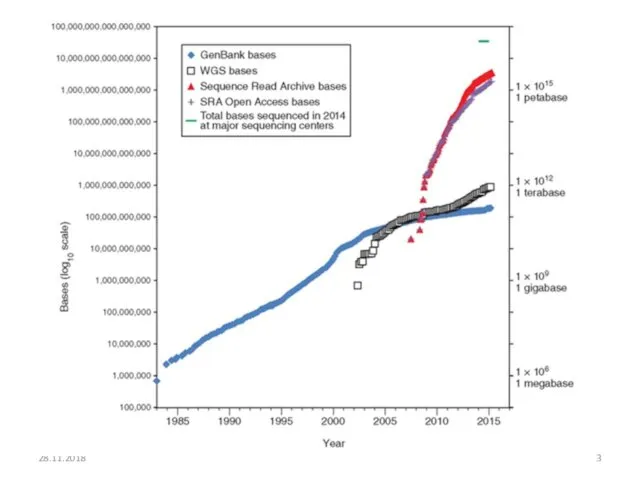
28.11.2018
28.11.2018
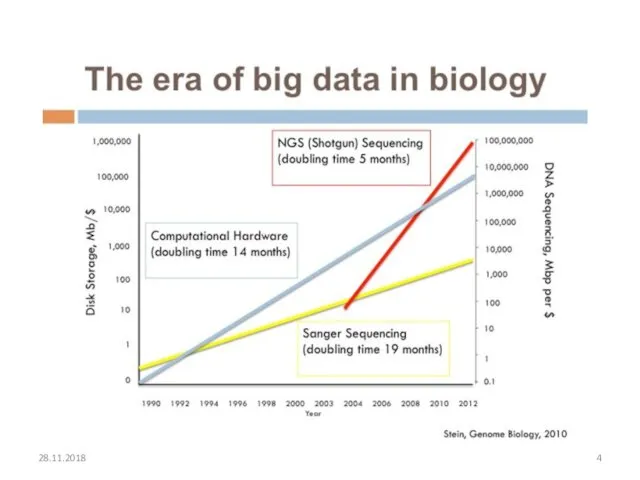
28.11.2018
28.11.2018
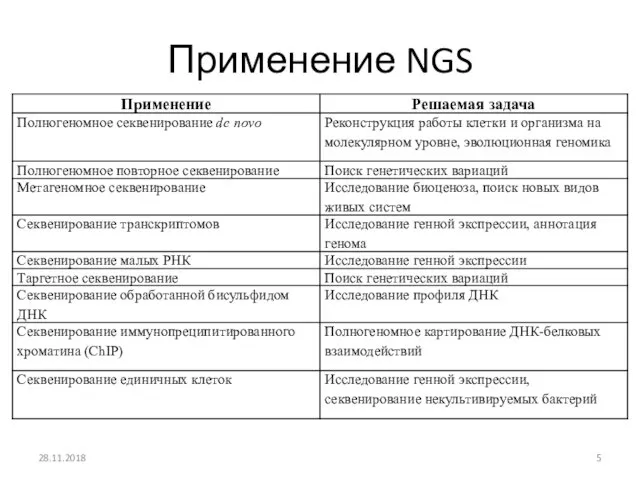
Применение NGS
28.11.2018
Применение NGS
28.11.2018

Основные термины
Геномные библиотеки - это коллекция геномной ДНК полученная от одного
Основные термины
Геномные библиотеки - это коллекция геномной ДНК полученная от одного
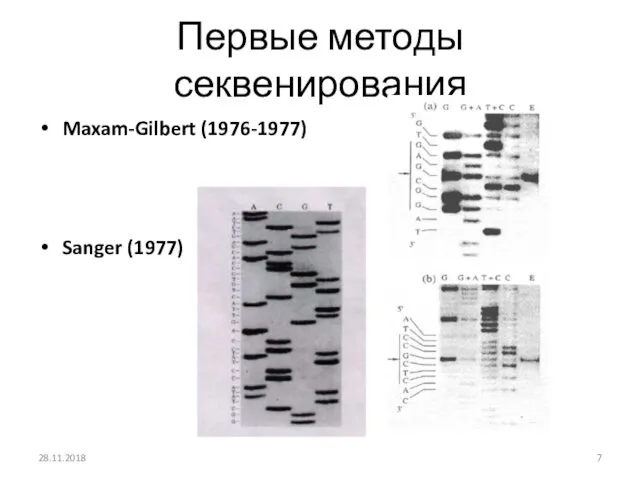
Первые методы секвенирования
Maxam-Gilbert (1976-1977)
Sanger (1977)
28.11.2018
Первые методы секвенирования
Maxam-Gilbert (1976-1977)
Sanger (1977)
28.11.2018
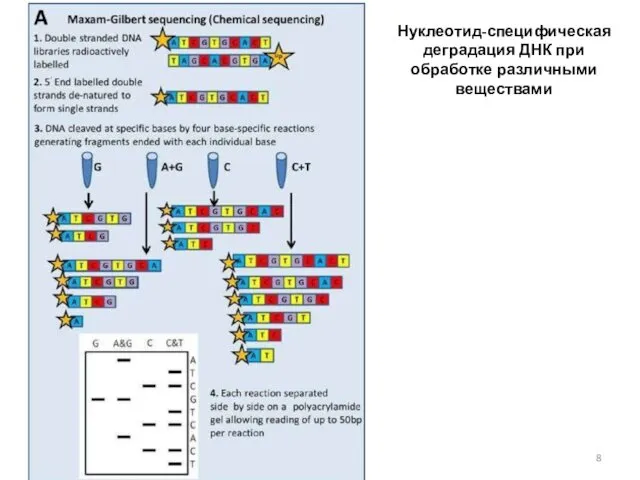
28.11.2018
Нуклеотид-специфическая деградация ДНК при обработке различными веществами
28.11.2018
Нуклеотид-специфическая деградация ДНК при обработке различными веществами
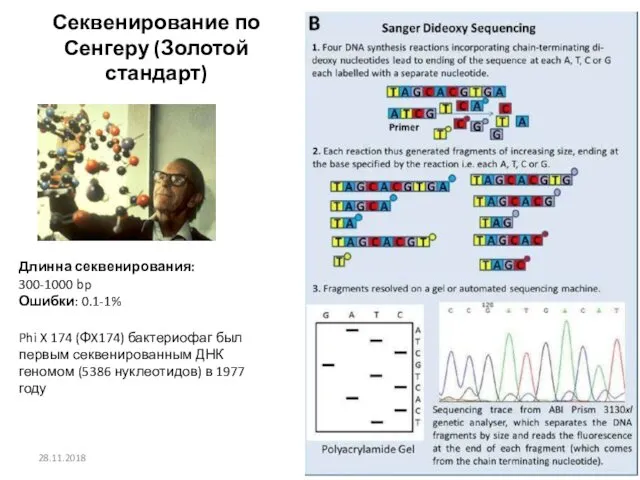
Секвенирование по Сенгеру (Золотой стандарт)
28.11.2018
Phi X 174 (ΦX174) бактериофаг был первым
Секвенирование по Сенгеру (Золотой стандарт)
28.11.2018
Phi X 174 (ΦX174) бактериофаг был первым
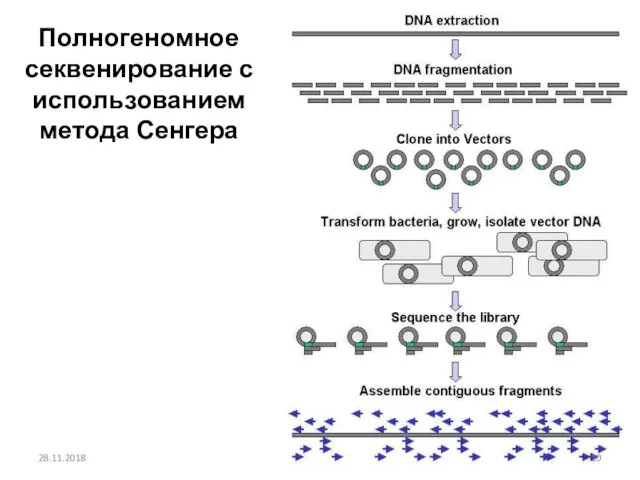
Полногеномное секвенирование с использованием метода Сенгера
28.11.2018
Полногеномное секвенирование с использованием метода Сенгера
28.11.2018

Проект геном человека
Размер генома – 3.2 Гб
Длительность – 10 лет
1990 –
Проект геном человека
Размер генома – 3.2 Гб
Длительность – 10 лет
1990 –

Секвенирование по Сенгеру
Плюсы:
Относительно низкий уровень ошибок
Удобное и дешевое секвенирование небольших фрагментов
Секвенирование по Сенгеру
Плюсы:
Относительно низкий уровень ошибок
Удобное и дешевое секвенирование небольших фрагментов

28.11.2018
28.11.2018

New Generation Sequencing
28.11.2018
Плюсы:
Простая подготовка ДНК библиотек (пробоподготовка)
Высокая производительность
Низкая стоимость секвенирования
Минусы:
Короткие риды
Относительно
New Generation Sequencing
28.11.2018
Плюсы:
Простая подготовка ДНК библиотек (пробоподготовка)
Высокая производительность
Низкая стоимость секвенирования
Минусы:
Короткие риды
Относительно
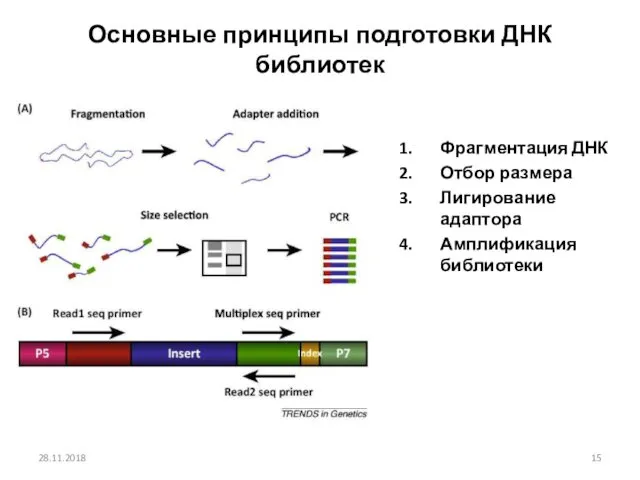
Основные принципы подготовки ДНК библиотек
Фрагментация ДНК
Отбор размера
Лигирование адаптора
Амплификация библиотеки
28.11.2018
Основные принципы подготовки ДНК библиотек
Фрагментация ДНК
Отбор размера
Лигирование адаптора
Амплификация библиотеки
28.11.2018
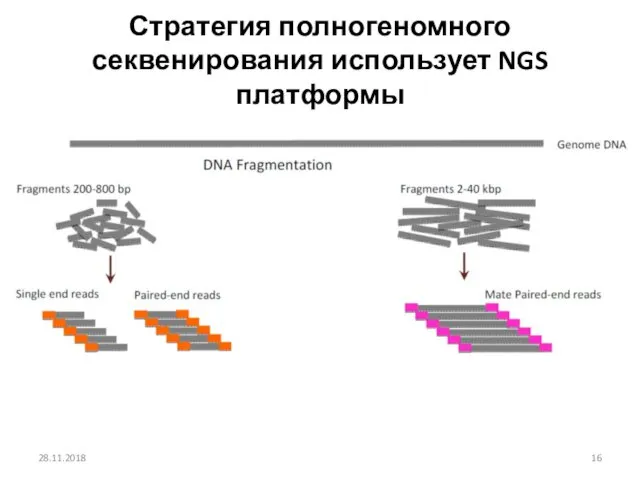
Стратегия полногеномного секвенирования использует NGS платформы
28.11.2018
Стратегия полногеномного секвенирования использует NGS платформы
28.11.2018
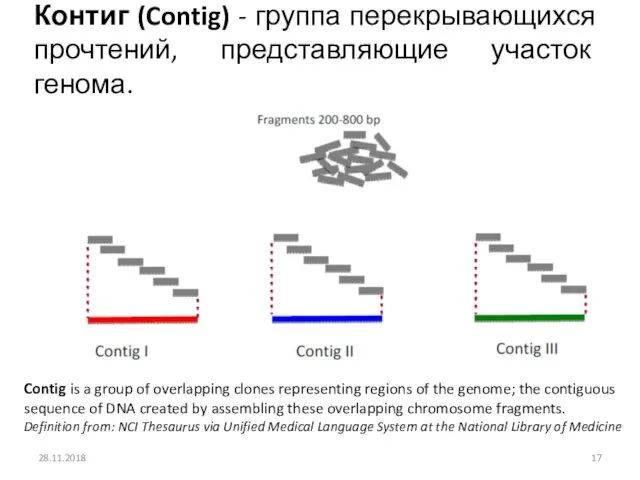
Контиг (Contig) - группа перекрывающихся прочтений, представляющие участок генома.
28.11.2018
Contig is a
Контиг (Contig) - группа перекрывающихся прочтений, представляющие участок генома.
28.11.2018
Contig is a
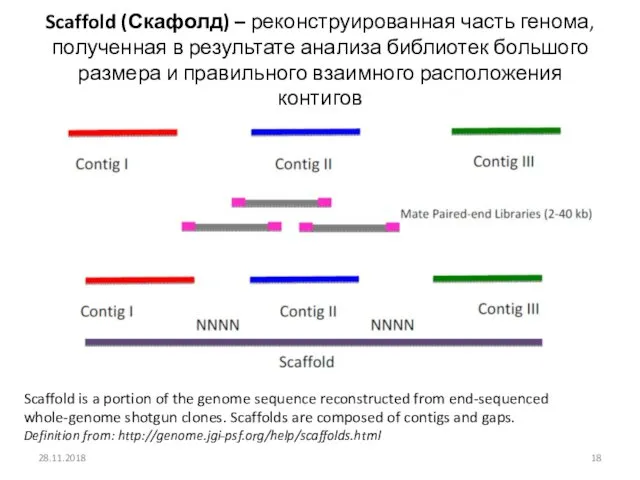
Scaffold (Скафолд) – реконструированная часть генома, полученная в результате анализа библиотек
Scaffold (Скафолд) – реконструированная часть генома, полученная в результате анализа библиотек
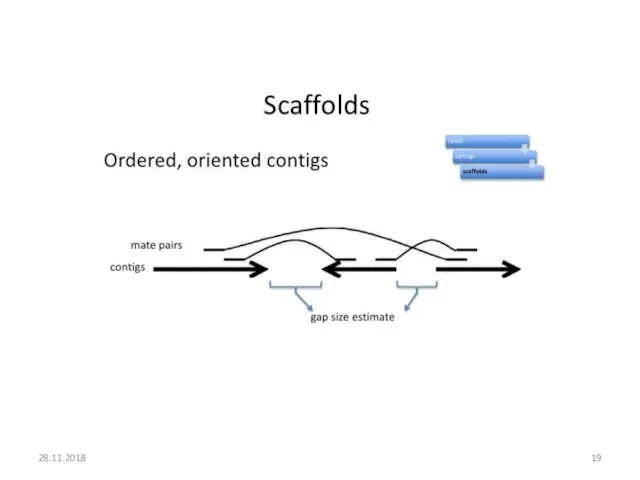
28.11.2018
28.11.2018
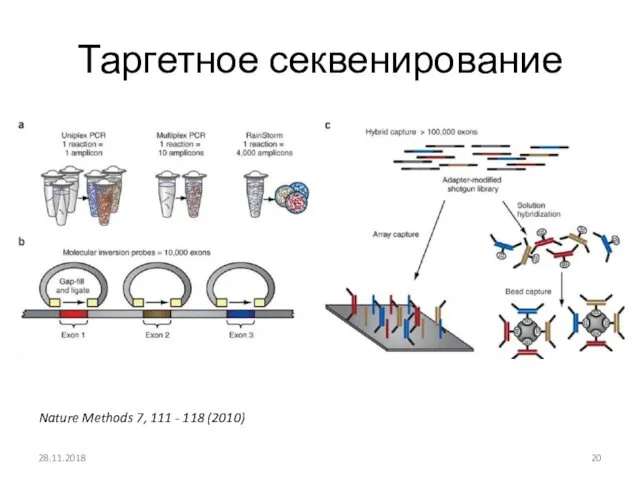
Таргетное секвенирование
28.11.2018
Nature Methods 7, 111 - 118 (2010)
Таргетное секвенирование
28.11.2018
Nature Methods 7, 111 - 118 (2010)

Индексирование (Баркодинг)
Можно за один запуск секвенатора прочитать несколько геномов или геномных
Индексирование (Баркодинг)
Можно за один запуск секвенатора прочитать несколько геномов или геномных
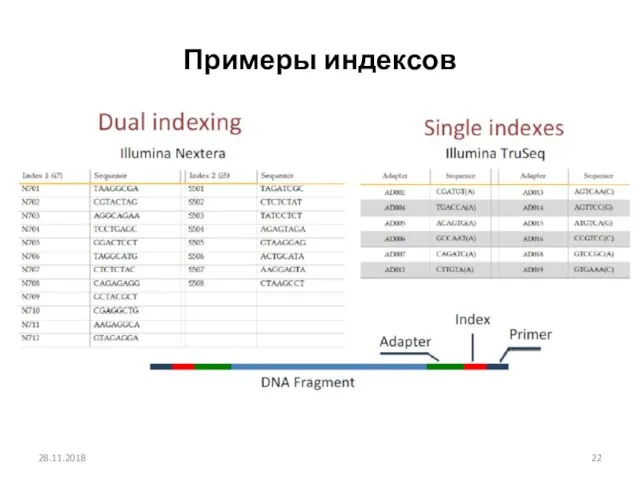
Примеры индексов
28.11.2018
Примеры индексов
28.11.2018

Платформы
28.11.2018
Платформы
28.11.2018

28.11.2018
28.11.2018

454 Sequencing Technology
28.11.2018
Фрагментация ДНК
Подготовка библиотеки
Пришивание адапторов к молекулам ДНК с двух
454 Sequencing Technology
28.11.2018
Фрагментация ДНК
Подготовка библиотеки
Пришивание адапторов к молекулам ДНК с двух

28.11.2018
Один фрагмент = одна бусина (bead)
Библиотека фрагментов ДНК прикрепляется к бусинам
28.11.2018
Один фрагмент = одна бусина (bead)
Библиотека фрагментов ДНК прикрепляется к бусинам
28.11.2018
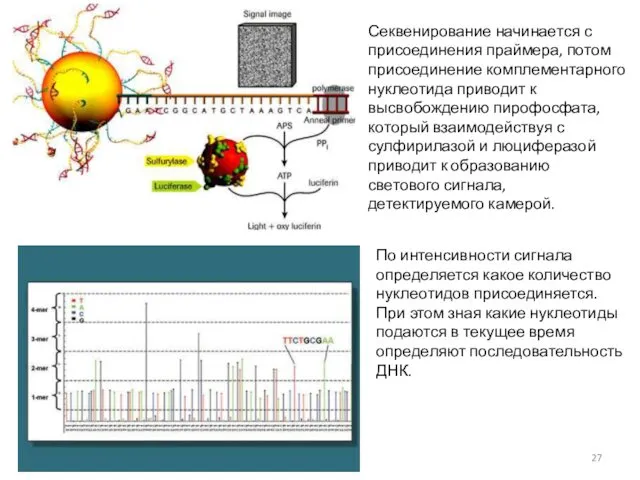
Секвенирование начинается с присоединения праймера, потом присоединение комплементарного нуклеотида приводит к
28.11.2018
Секвенирование начинается с присоединения праймера, потом присоединение комплементарного нуклеотида приводит к

28.11.2018
28.11.2018

Ion Torrent
Подготовка библиотеки похожа на Roche 454
• фрагментация ДНК
• Прикрепление адаптера
•
Ion Torrent
Подготовка библиотеки похожа на Roche 454
• фрагментация ДНК
• Прикрепление адаптера
•
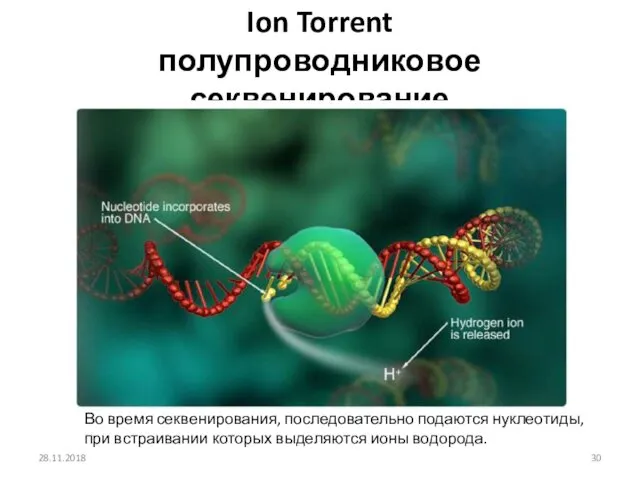
Ion Torrent
полупроводниковое секвенирование
28.11.2018
Во время секвенирования, последовательно подаются нуклеотиды, при встраивании
Ion Torrent
полупроводниковое секвенирование
28.11.2018
Во время секвенирования, последовательно подаются нуклеотиды, при встраивании
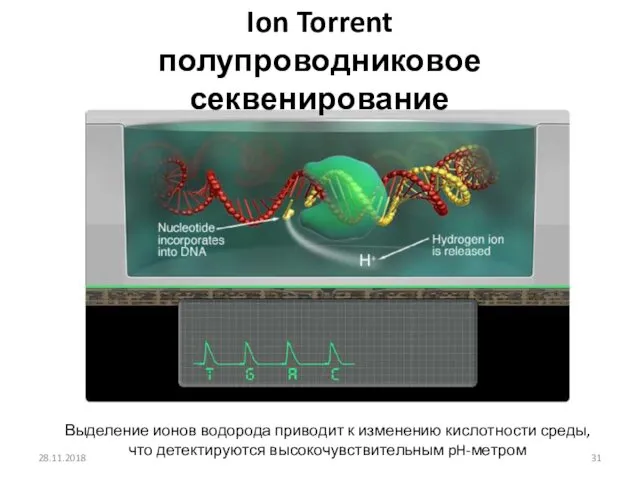
28.11.2018
Ion Torrent
полупроводниковое секвенирование
Выделение ионов водорода приводит к изменению кислотности среды,
28.11.2018
Ion Torrent
полупроводниковое секвенирование
Выделение ионов водорода приводит к изменению кислотности среды,
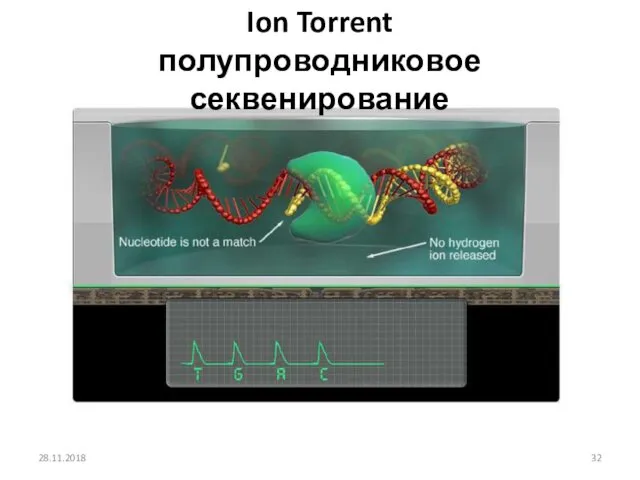
28.11.2018
Ion Torrent
полупроводниковое секвенирование
28.11.2018
Ion Torrent
полупроводниковое секвенирование
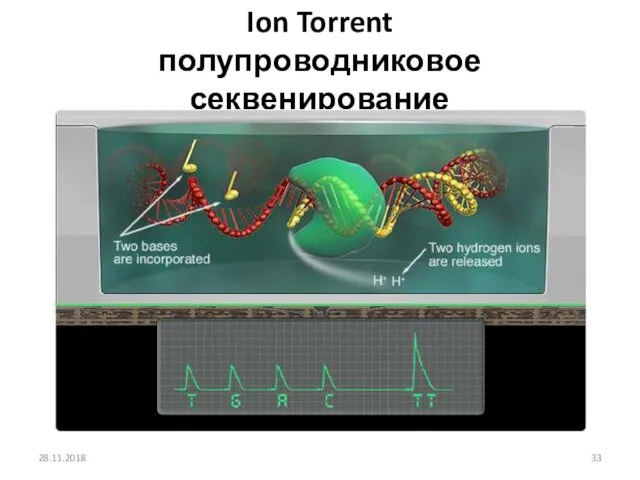
28.11.2018
Ion Torrent
полупроводниковое секвенирование
28.11.2018
Ion Torrent
полупроводниковое секвенирование

28.11.2018
28.11.2018

SOLiD
28.11.2018
Подготовка библиотеки похожа на Roche 454
• фрагментация ДНК
• Прикрепление адаптера
• Эмульсионная
SOLiD
28.11.2018
Подготовка библиотеки похожа на Roche 454
• фрагментация ДНК
• Прикрепление адаптера
• Эмульсионная
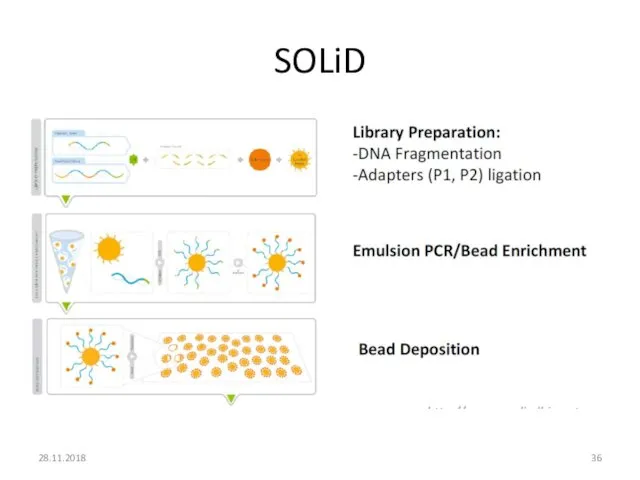
SOLiD
28.11.2018
SOLiD
28.11.2018
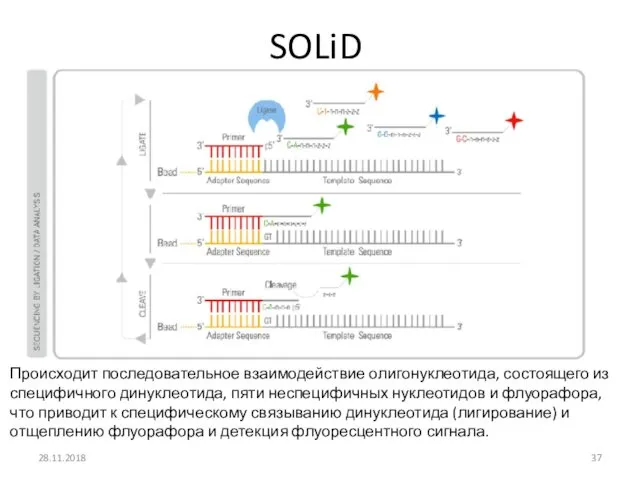
SOLiD
28.11.2018
Происходит последовательное взаимодействие олигонуклеотида, состоящего из специфичного динуклеотида, пяти неспецифичных нуклеотидов
SOLiD
28.11.2018
Происходит последовательное взаимодействие олигонуклеотида, состоящего из специфичного динуклеотида, пяти неспецифичных нуклеотидов
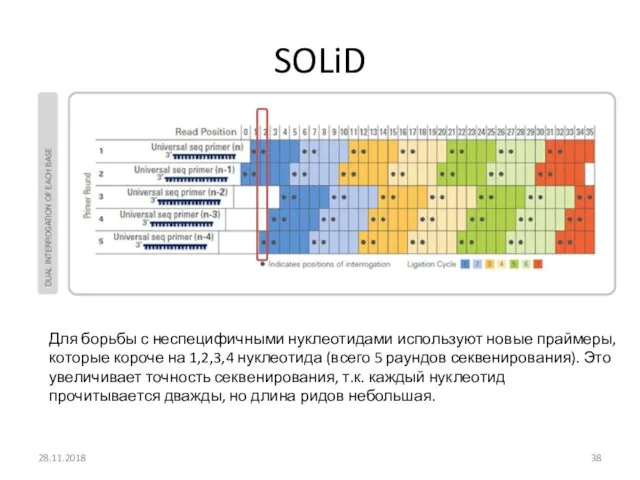
SOLiD
28.11.2018
Для борьбы с неспецифичными нуклеотидами используют новые праймеры, которые короче на
SOLiD
28.11.2018
Для борьбы с неспецифичными нуклеотидами используют новые праймеры, которые короче на
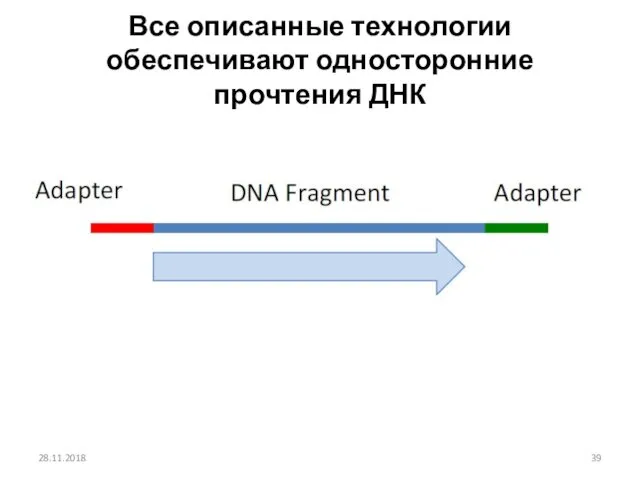
Все описанные технологии обеспечивают односторонние прочтения ДНК
28.11.2018
Все описанные технологии обеспечивают односторонние прочтения ДНК
28.11.2018

28.11.2018
28.11.2018
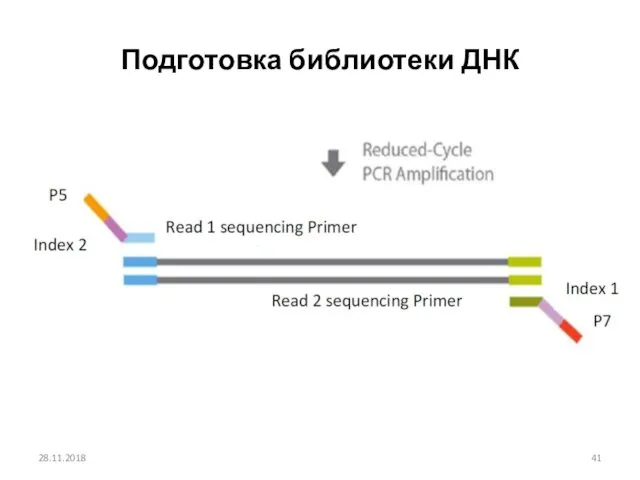
Подготовка библиотеки ДНК
28.11.2018
Подготовка библиотеки ДНК
28.11.2018

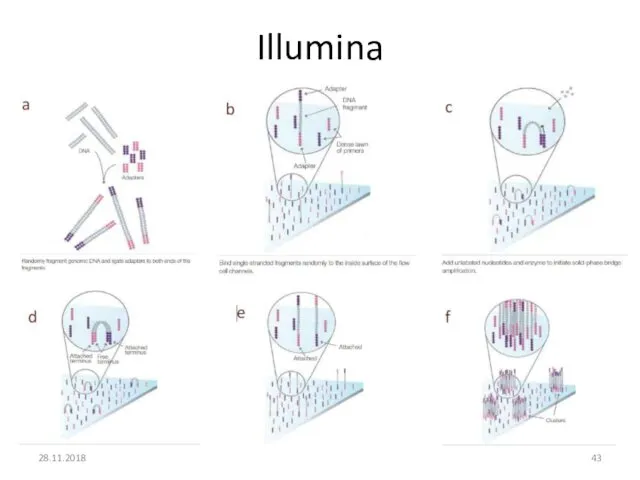
Illumina
Гибридизация ДНК-библиотек
Генерация кластеров (ПЦР)
Секвенирование синтезом
28.11.2018
http://www.youtube.com/watch?v=HMyCqWhwB8E
Illumina
Гибридизация ДНК-библиотек
Генерация кластеров (ПЦР)
Секвенирование синтезом
28.11.2018
http://www.youtube.com/watch?v=HMyCqWhwB8E
28.11.2018
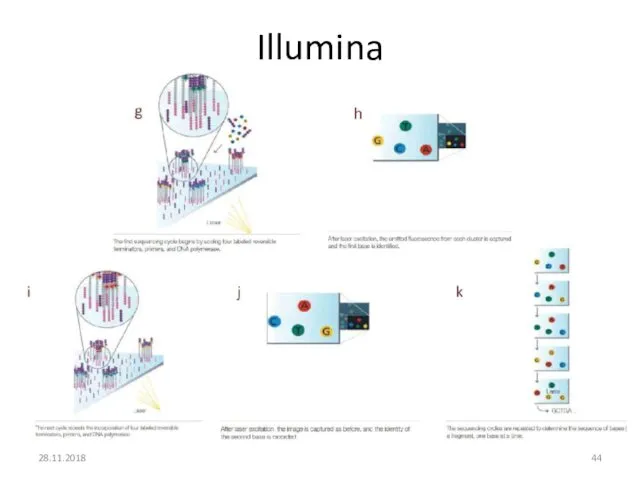
Illumina
28.11.2018
Illumina
28.11.2018
Illumina
28.11.2018
Illumina

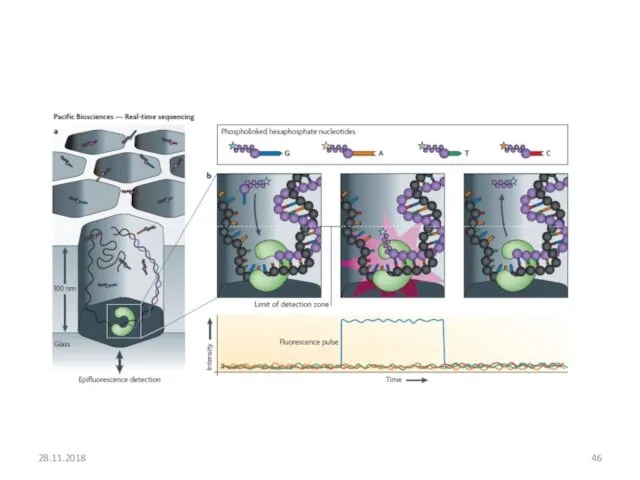
Pacific Biosciences
single molecule real-time (SMRT) sequencing
Одномолекулярное секвенирование в реальном времени
Секвенировании без
Pacific Biosciences
single molecule real-time (SMRT) sequencing
Одномолекулярное секвенирование в реальном времени
Секвенировании без
28.11.2018
28.11.2018

28.11.2018
28.11.2018
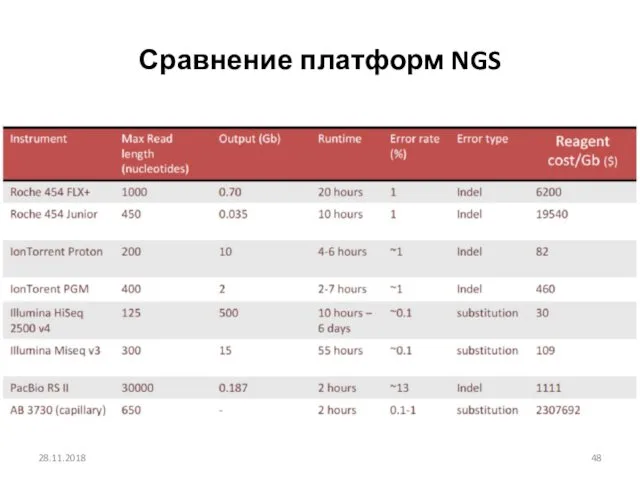
Сравнение платформ NGS
28.11.2018
Сравнение платформ NGS
28.11.2018
Контроль качества данных
28.11.2018
Контроль качества данных
28.11.2018

28.11.2018
28.11.2018
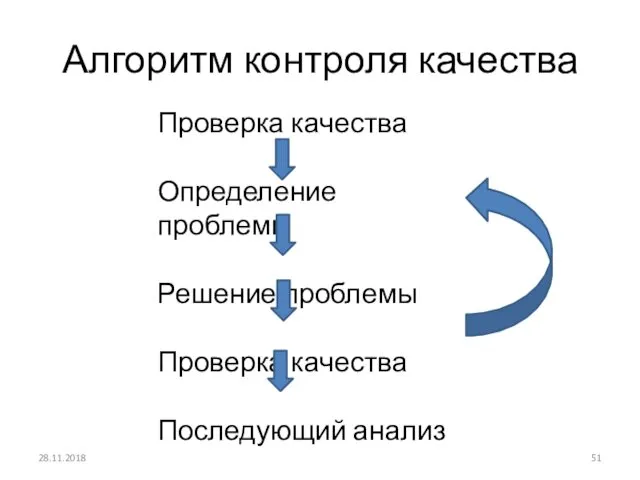
Алгоритм контроля качества
28.11.2018
Проверка качества
Определение проблемы
Решение проблемы
Проверка качества
Последующий анализ
Алгоритм контроля качества
28.11.2018
Проверка качества
Определение проблемы
Решение проблемы
Проверка качества
Последующий анализ

Зачем чистить данные?
28.11.2018
• Риды низкого качества
• Контаминация (примесь ДНК другого организма)
•
Зачем чистить данные?
28.11.2018
• Риды низкого качества
• Контаминация (примесь ДНК другого организма)
•

FASTA и FASTQ форматы
28.11.2018
FASTA
FASTQ
Линия начинающаяся с @ содержит идентификатор
FASTA и FASTQ форматы
28.11.2018
FASTA
FASTQ
Линия начинающаяся с @ содержит идентификатор
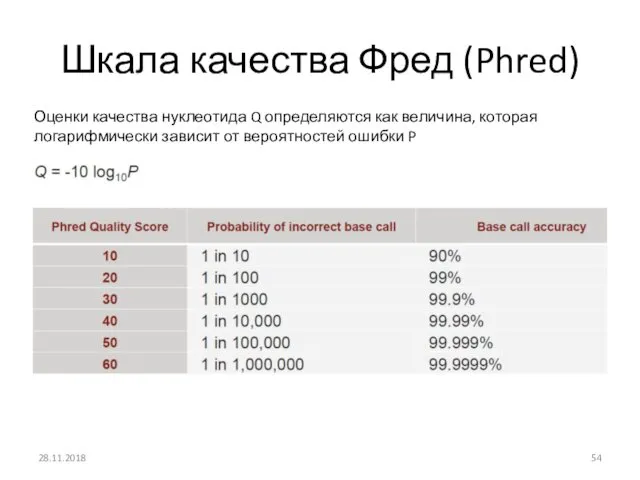
Шкала качества Фред (Phred)
28.11.2018
Оценки качества нуклеотида Q определяются как величина, которая
Шкала качества Фред (Phred)
28.11.2018
Оценки качества нуклеотида Q определяются как величина, которая
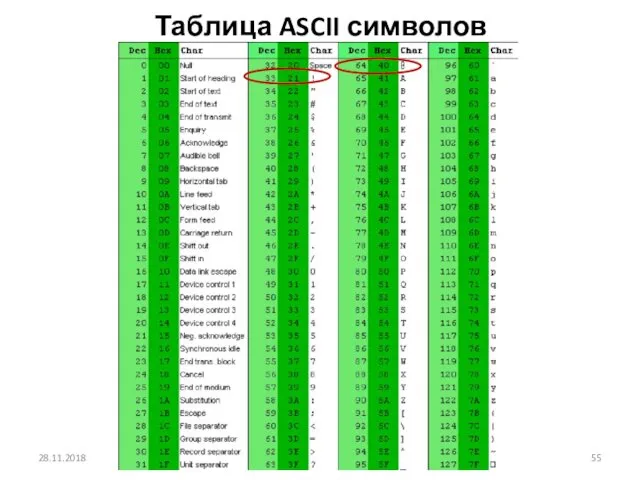
Таблица ASCII символов
28.11.2018
Таблица ASCII символов
28.11.2018

Разные Phred шкалы
28.11.2018
Разные Phred шкалы
28.11.2018
Cборка генома
28.11.2018
Cборка генома
28.11.2018
28.11.2018
28.11.2018

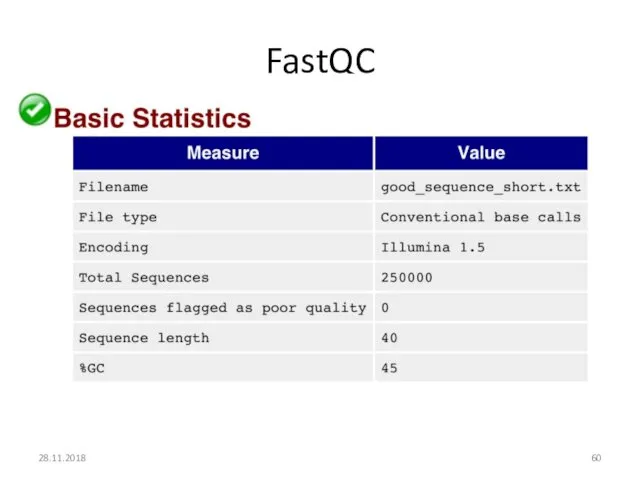
FastQC – инструмент для контроля качества данных
На вход – исходные данные
FastQC – инструмент для контроля качества данных
На вход – исходные данные
FastQC
28.11.2018
FastQC
28.11.2018
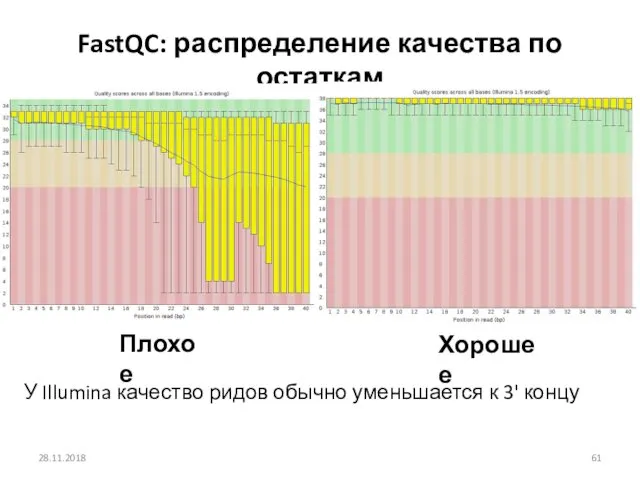
FastQC: распределение качества по остаткам
28.11.2018
Плохое
Хорошее
У Illumina качество ридов обычно уменьшается к
FastQC: распределение качества по остаткам
28.11.2018
Плохое
Хорошее
У Illumina качество ридов обычно уменьшается к
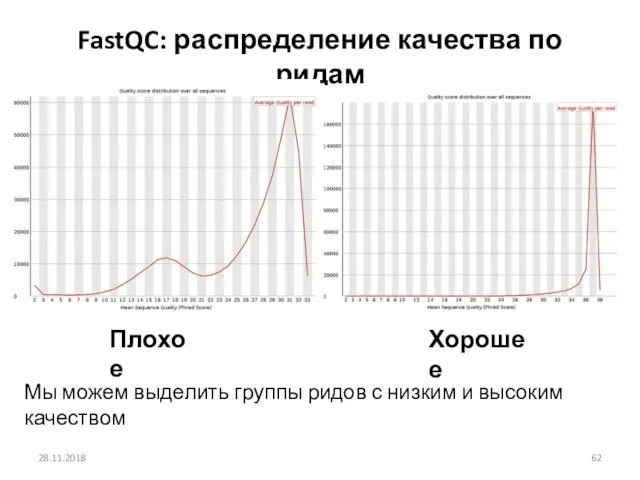
28.11.2018
FastQC: распределение качества по ридам
Плохое
Хорошее
Мы можем выделить группы ридов с низким
28.11.2018
FastQC: распределение качества по ридам
Плохое
Хорошее
Мы можем выделить группы ридов с низким
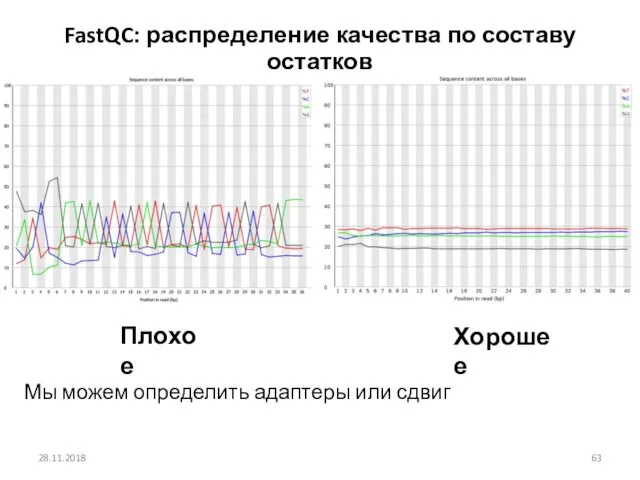
28.11.2018
FastQC: распределение качества по составу остатков
Плохое
Хорошее
Мы можем определить адаптеры или сдвиг
28.11.2018
FastQC: распределение качества по составу остатков
Плохое
Хорошее
Мы можем определить адаптеры или сдвиг
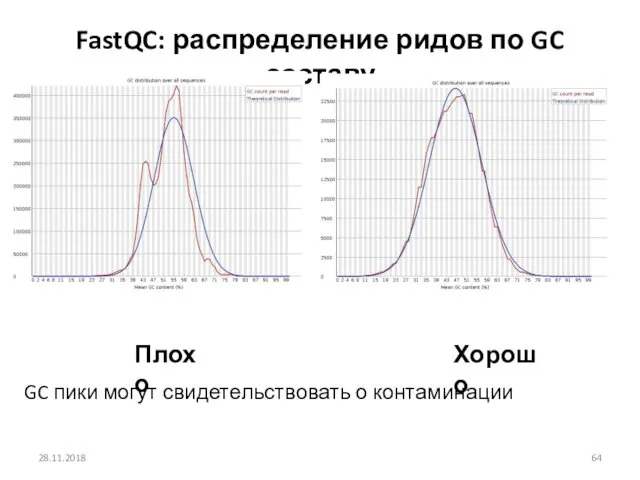
28.11.2018
FastQC: распределение ридов по GC составу
Плохо
Хорошо
GC пики могут свидетельствовать о контаминации
28.11.2018
FastQC: распределение ридов по GC составу
Плохо
Хорошо
GC пики могут свидетельствовать о контаминации
28.11.2018
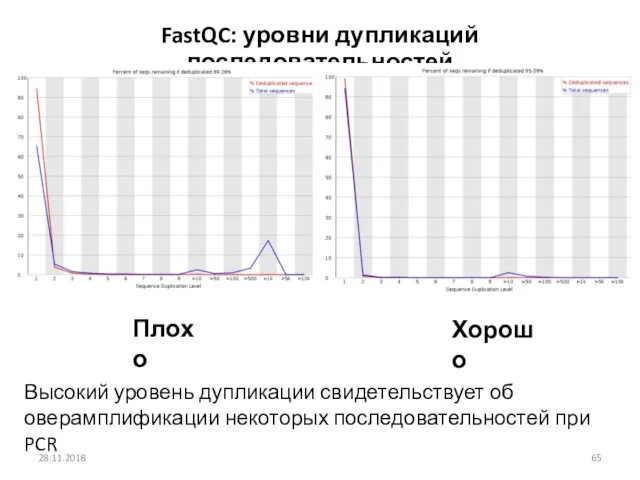
FastQC: уровни дупликаций последовательностей
Плохо
Хорошо
Высокий уровень дупликации свидетельствует об оверамплификации некоторых последовательностей
28.11.2018
FastQC: уровни дупликаций последовательностей
Плохо
Хорошо
Высокий уровень дупликации свидетельствует об оверамплификации некоторых последовательностей
28.11.2018
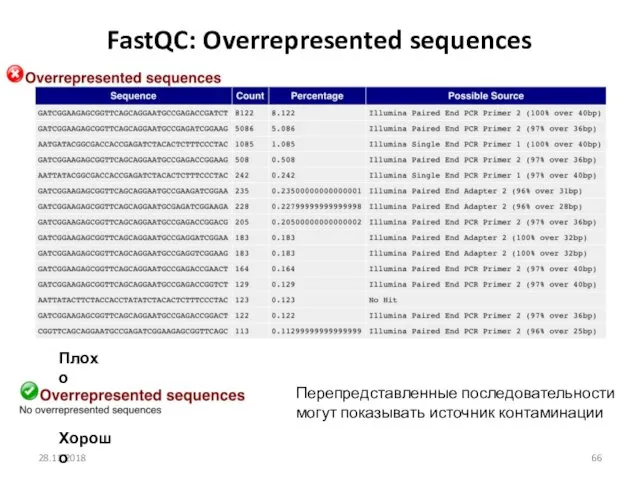
FastQC: Overrepresented sequences
Плохо
Хорошо
Перепредставленные последовательности могут показывать источник контаминации
28.11.2018
FastQC: Overrepresented sequences
Плохо
Хорошо
Перепредставленные последовательности могут показывать источник контаминации

28.11.2018
FastQC: Качество ячеек
Плохо
Хорошо
У Illumina можно определить проблемы с ячейками
28.11.2018
FastQC: Качество ячеек
Плохо
Хорошо
У Illumina можно определить проблемы с ячейками

Шаги препроцессинга
Фильтрация данных по качеству
Удаление ридов, качество которых ниже определенного порога;
Обрезание
Шаги препроцессинга
Фильтрация данных по качеству
Удаление ридов, качество которых ниже определенного порога;
Обрезание

У нас есть очищенные данные. Что дальше?
Сборка de novo
Сборка по референсному
У нас есть очищенные данные. Что дальше?
Сборка de novo
Сборка по референсному
Сборка de novo
28.11.2018
Возьмем большое количество коротких секвенированных ридов и поместим их
Сборка de novo
28.11.2018
Возьмем большое количество коротких секвенированных ридов и поместим их
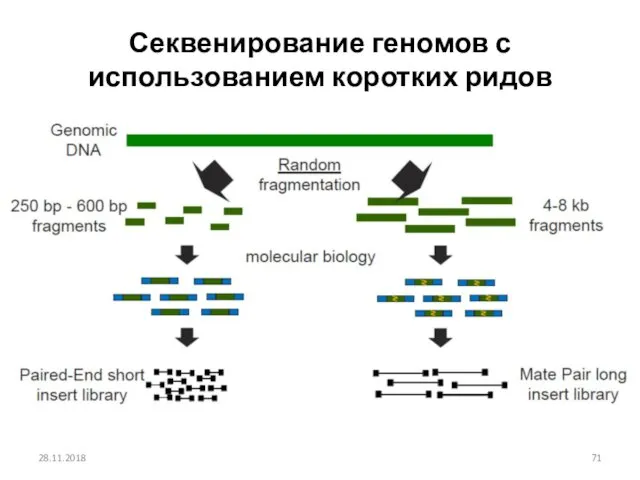
Секвенирование геномов с использованием коротких ридов
28.11.2018
Секвенирование геномов с использованием коротких ридов
28.11.2018
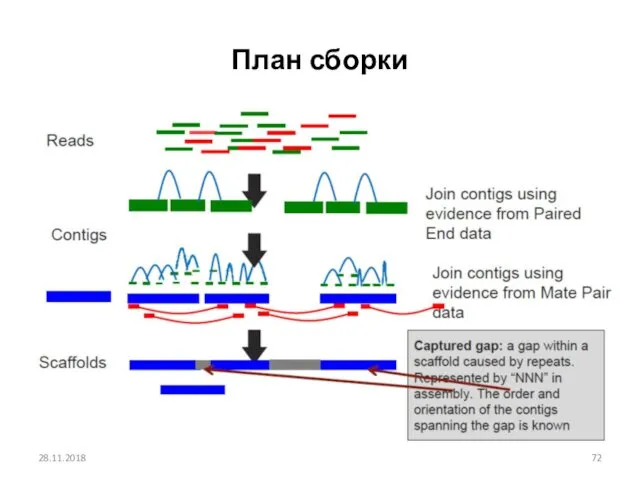
План сборки
28.11.2018
План сборки
28.11.2018
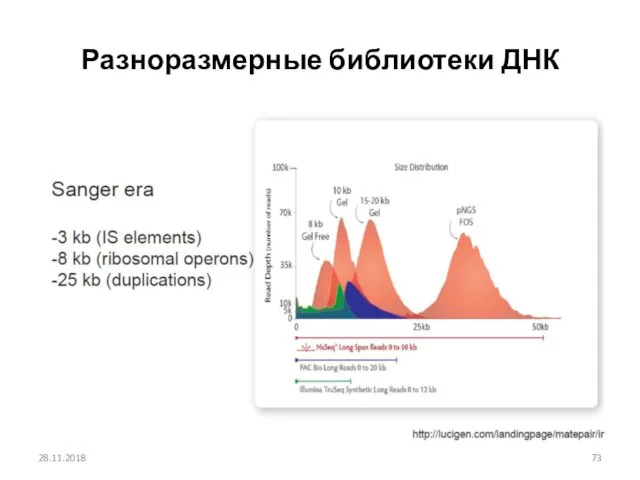
Разноразмерные библиотеки ДНК
28.11.2018
Разноразмерные библиотеки ДНК
28.11.2018
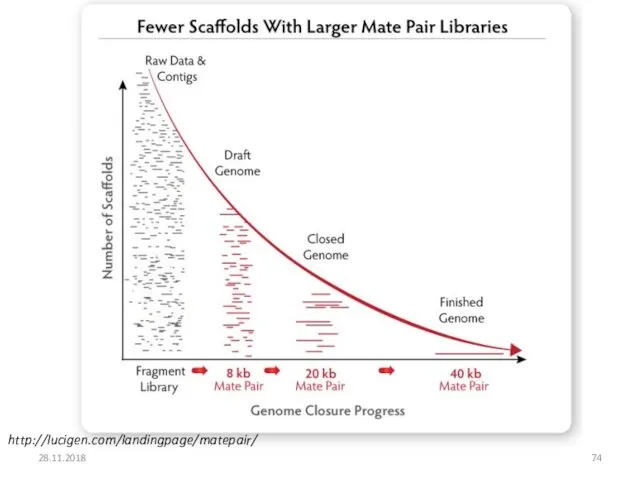
28.11.2018
http://lucigen.com/landingpage/matepair/
28.11.2018
http://lucigen.com/landingpage/matepair/
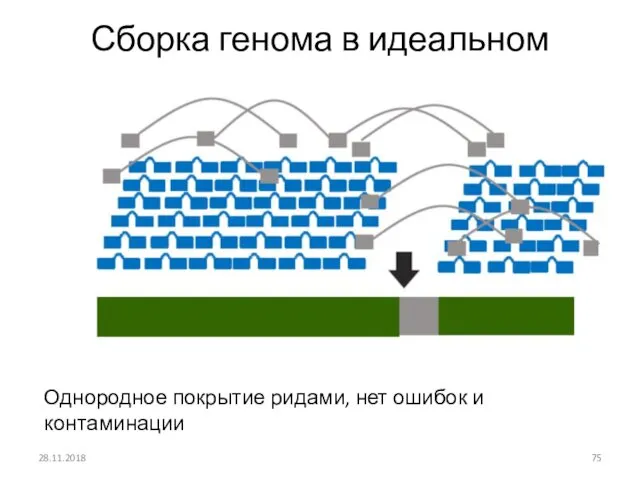
Сборка генома в идеальном случае
28.11.2018
Однородное покрытие ридами, нет ошибок и контаминации
Сборка генома в идеальном случае
28.11.2018
Однородное покрытие ридами, нет ошибок и контаминации
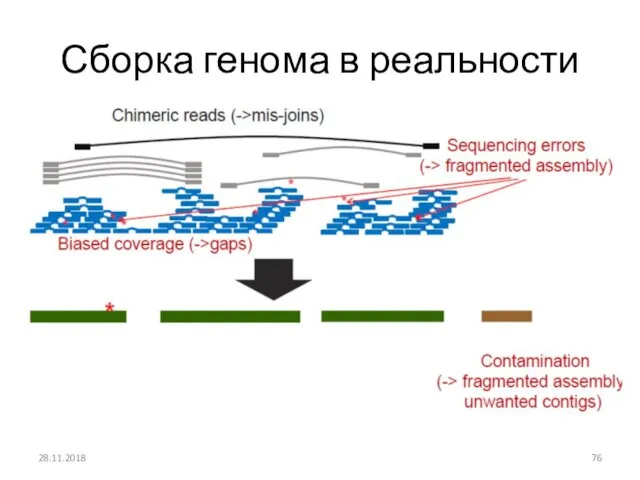
Сборка генома в реальности
28.11.2018
Сборка генома в реальности
28.11.2018

28.11.2018
Кафедра биоинформатики МБФ РНИМУ
28.11.2018
Кафедра биоинформатики МБФ РНИМУ

Выбор правильной программы - сборщика геномов (ассемблер)
На сколько большой геном?
Существуют ли
Выбор правильной программы - сборщика геномов (ассемблер)
На сколько большой геном?
Существуют ли
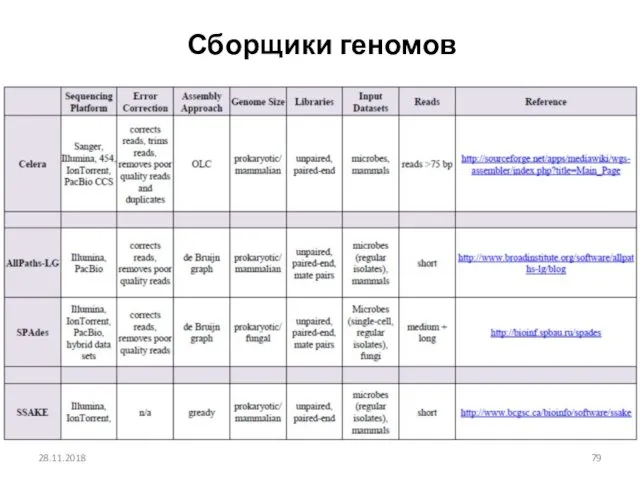
Сборщики геномов
28.11.2018
Сборщики геномов
28.11.2018

Оценка качества сборки генома
Количество контигов
Общая длинна всех контигов
Длинна наибольшего контига
Количество неправильно
Оценка качества сборки генома
Количество контигов
Общая длинна всех контигов
Длинна наибольшего контига
Количество неправильно
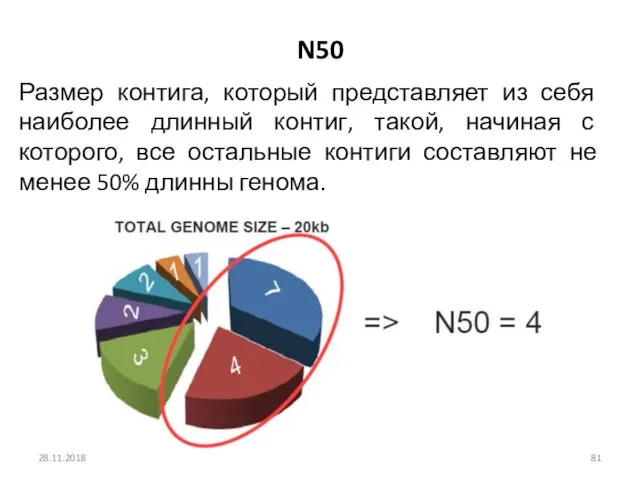
N50
Размер контига, который представляет из себя наиболее длинный контиг, такой, начиная
N50
Размер контига, который представляет из себя наиболее длинный контиг, такой, начиная
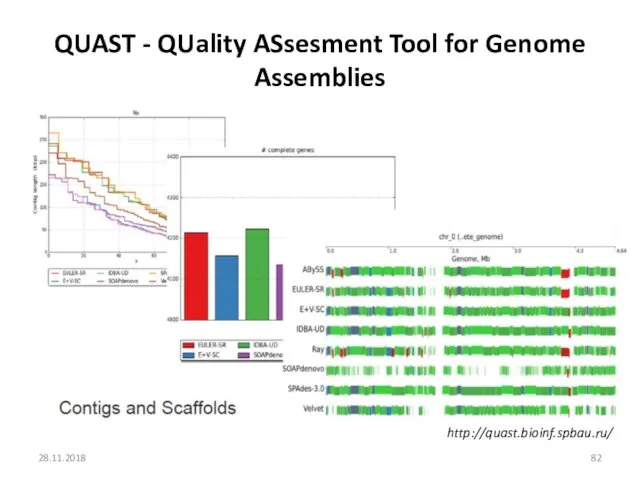
QUAST - QUality ASsesment Tool for Genome Assemblies
28.11.2018
http://quast.bioinf.spbau.ru/
QUAST - QUality ASsesment Tool for Genome Assemblies
28.11.2018
http://quast.bioinf.spbau.ru/

28.11.2018
28.11.2018

Реальные графы де Брюйна
28.11.2018
Реальные графы де Брюйна
28.11.2018
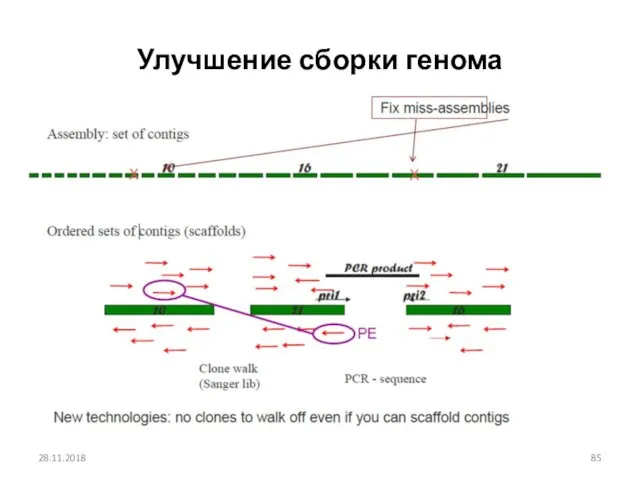
Улучшение сборки генома
28.11.2018
Улучшение сборки генома
28.11.2018
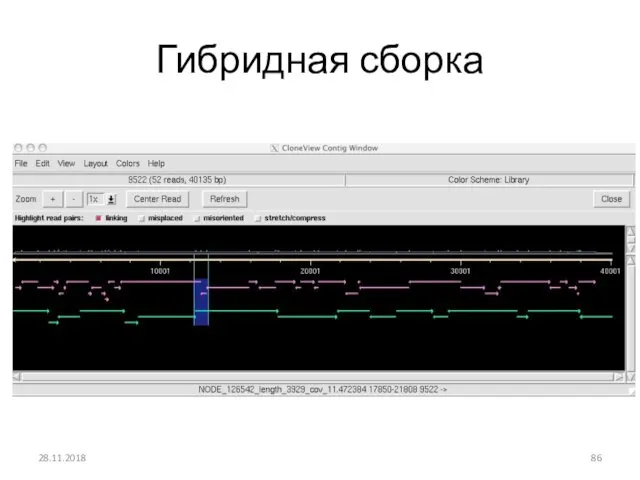
Гибридная сборка
28.11.2018
Гибридная сборка
28.11.2018
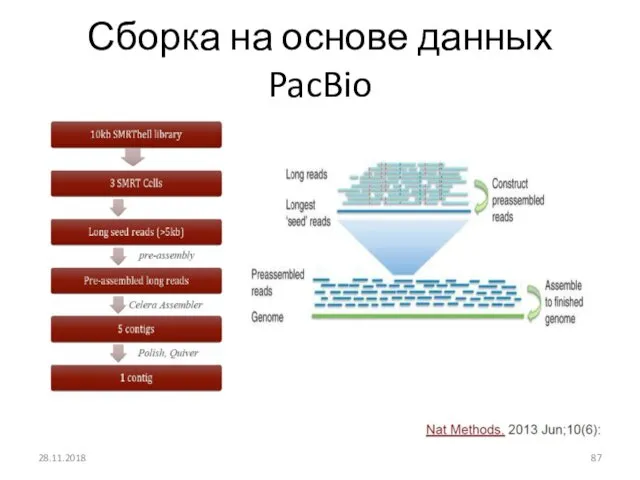
Сборка на основе данных PacBio
28.11.2018
Сборка на основе данных PacBio
28.11.2018
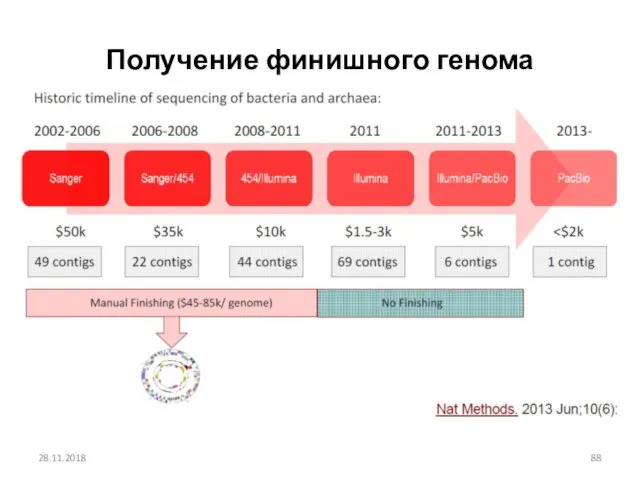
Получение финишного генома
28.11.2018
Получение финишного генома
28.11.2018

Зачем нужны финишные геномы?
Функциональные геномные исследования требуют высококачественной, полной последовательности генома
Зачем нужны финишные геномы?
Функциональные геномные исследования требуют высококачественной, полной последовательности генома
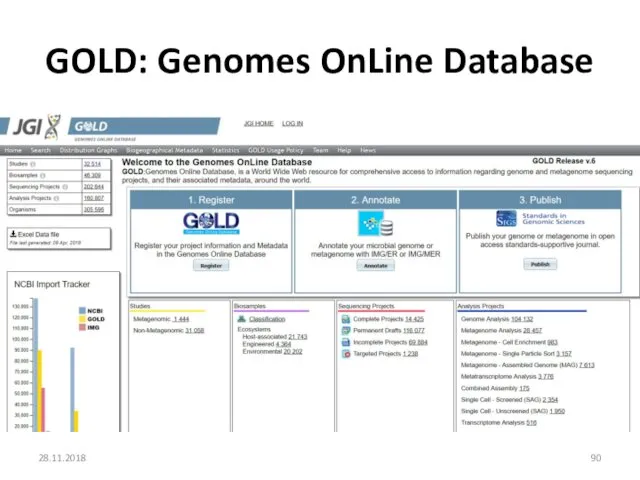
GOLD: Genomes OnLine Database
28.11.2018
GOLD: Genomes OnLine Database
28.11.2018
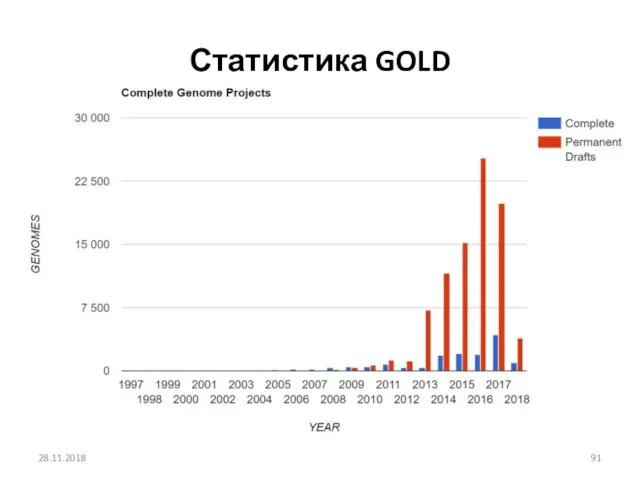
Статистика GOLD
28.11.2018
Статистика GOLD
28.11.2018
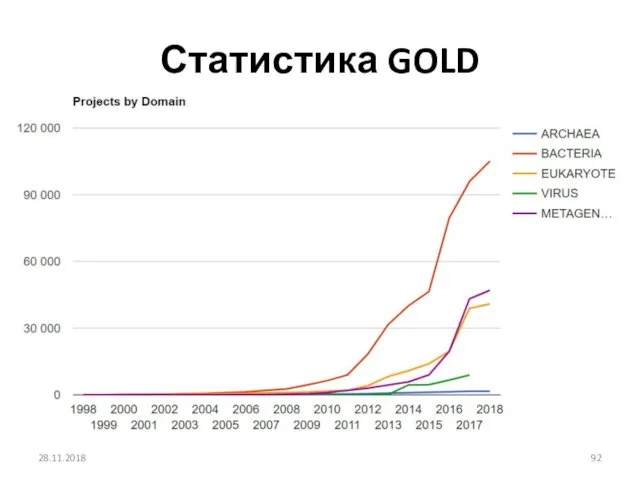
Статистика GOLD
28.11.2018
Статистика GOLD
28.11.2018
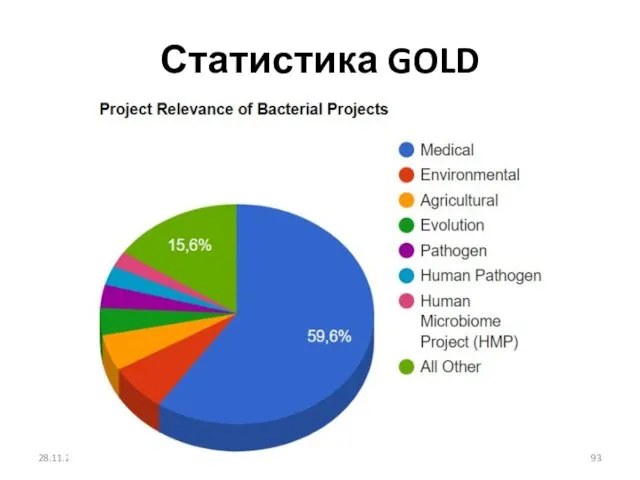
Статистика GOLD
28.11.2018
Статистика GOLD
28.11.2018
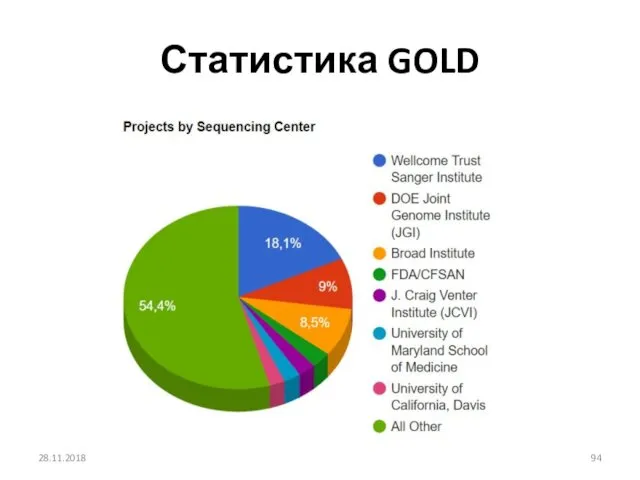
Статистика GOLD
28.11.2018
Статистика GOLD
28.11.2018
NCBI Genome
28.11.2018
NCBI Genome
28.11.2018
NCBI Genome
28.11.2018
NCBI Genome
28.11.2018
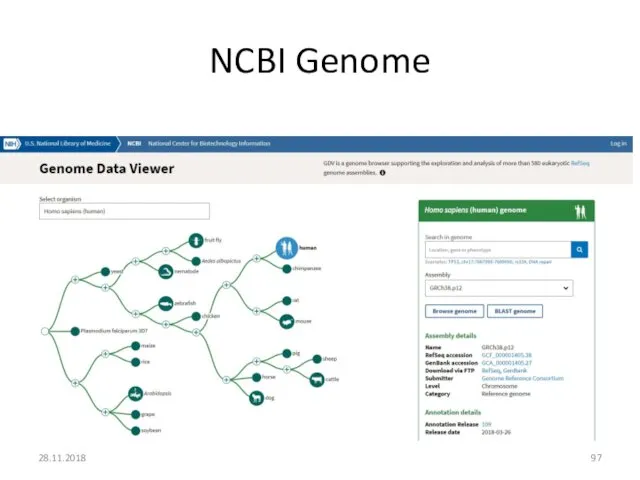
NCBI Genome
28.11.2018
NCBI Genome
28.11.2018
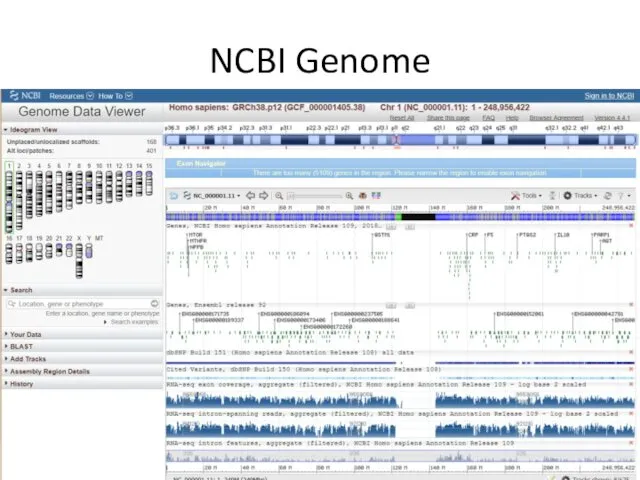
NCBI Genome
28.11.2018
NCBI Genome
28.11.2018
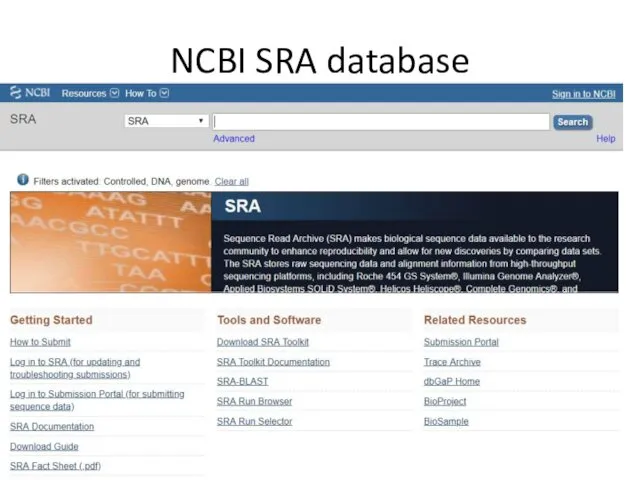
NCBI SRA database
28.11.2018
NCBI SRA database
28.11.2018
 Все о картофеле
Все о картофеле Орган слуха
Орган слуха Жилкування листків
Жилкування листків Кисломолочные бактерии. 5 класс
Кисломолочные бактерии. 5 класс Мимические мышцы лица
Мимические мышцы лица Интересные факты про бобра
Интересные факты про бобра Огород без химии, или бюджетные удобрения
Огород без химии, или бюджетные удобрения Матричные биосинтезы
Матричные биосинтезы Класс Пресмыкающиеся, или Рептилии. Отряд Чешуйчатые
Класс Пресмыкающиеся, или Рептилии. Отряд Чешуйчатые Класс Паукообразные
Класс Паукообразные Эпизоотологическое и эпидемиологическое значение грызунов. Дератизация
Эпизоотологическое и эпидемиологическое значение грызунов. Дератизация Биохимия нервной ткани
Биохимия нервной ткани Хищные птицы Республики Коми
Хищные птицы Республики Коми Открытый урок. Генетика как наука
Открытый урок. Генетика как наука В1 витамині
В1 витамині Визначення типу шкіри на різних ділянках обличчя та складання правил догляду за власною шкірою
Визначення типу шкіри на різних ділянках обличчя та складання правил догляду за власною шкірою Ауыз қуысы. Ауыз қуысы мүшелері және оның паталогиясы
Ауыз қуысы. Ауыз қуысы мүшелері және оның паталогиясы Тестовая работа по теме Корень. Строение корня, 6 класс.
Тестовая работа по теме Корень. Строение корня, 6 класс. Теории происхождения человека
Теории происхождения человека Цветковые (покрытосемянные) растения
Цветковые (покрытосемянные) растения Пагін і його будова
Пагін і його будова Презентация Технология Развитие критического мышления через чтение и письмо - способ формирования системы универсальных учебных действий на уроках биологии.
Презентация Технология Развитие критического мышления через чтение и письмо - способ формирования системы универсальных учебных действий на уроках биологии. Бактерії і актиноміцети- як збудники хвороб рослин. Лекція №4
Бактерії і актиноміцети- як збудники хвороб рослин. Лекція №4 Клетка – основная структурная единица организма
Клетка – основная структурная единица организма Законы Менделя
Законы Менделя Височная кость. Каналы височной кости
Височная кость. Каналы височной кости Хвойные растения
Хвойные растения презентация на тему Плоды
презентация на тему Плоды