Аминқышқылы метаболизміндегі тұқым қуалайтын аурулар. Триптофан алмасуындағы бұзылыстар. Хартрап ауруы презентация
- Аминқышқылы метаболизміндегі тұқым қуалайтын аурулар. Триптофан алмасуындағы бұзылыстар. Хартрап ауруы

Содержание
- 2. Триптофан является одной из 8-ми незаменимых для взрослого человека аминокислот. Относится к ароматическим альфа-аминокислотам, химическое название:
- 4. Болезнь Хартнупа Болезнь Хартнупа — это редкое наследственное заболевание, в основе которого лежит нарушение обмена аминокислот
- 5. Этиопатогенез При синдроме Хартнупа (имя больного, родители которого были двоюродными братом и сестрой) в результате дефекта
- 6. Причиной данного заболевания является мутация гена, который отвечает за обмен аминокислот, в первую очередь за обмен
- 8. Кожа Чаще поражению подвергаются воздействию солнечных лучей, а именно кожа лица, шеи, тыл кистей и стоп,
- 12. пальцы рук и ног утолщаются, складки в области суставов сглаживаются. Ладони приобретают желтоватый оттенок, шелушатся; ногти
- 13. Факторы, способствующие развитию данного заболевания плохое питание (недоедание; употребление продуктов, в которых содержится мало животного белка,
- 14. Диагностика Общий осмотр — видны изменения: Сбор анамнеза (у родственников больного выясняется наличие случаев данного заболевания).
- 15. Лечение болезни Хартнупа Больному назначают длительный прием витамина РР (никотиновой кислоты) и иных витаминов группы В.
- 16. ЦИСТАТИОНИНУРИЯ (цистатионин -f- греческий uron моча) — наследственная аномалия обмена серосодержащих аминокислот, основным проявлением которой является
- 19. Клиническая картина характеризуется задержкой умственного развития ребенка. У некоторых больных с цистатионинурией выявляются тромбоцитопения, эндокринные нарушения
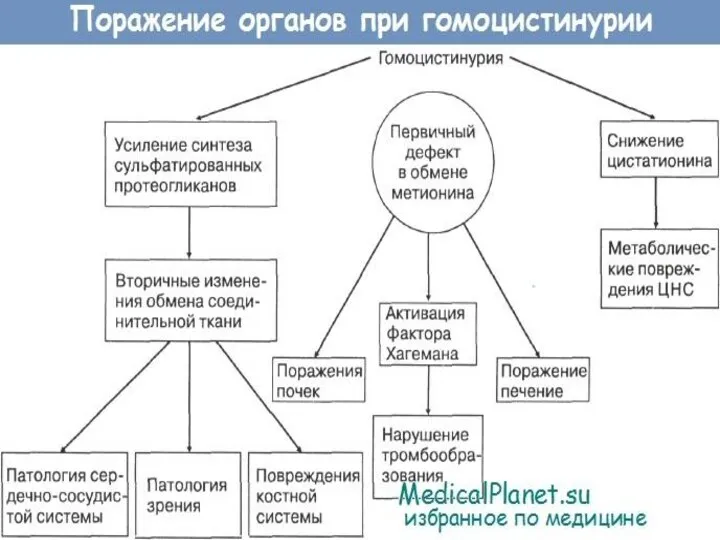
- 20. Гомоцистинурия Это наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот, в первую очередь метионина.
- 24. Установлено, что появление и увеличение концентрации гомоцистина в сыворотке крови способствует образованию некротически-дегенеративных участков в почках,
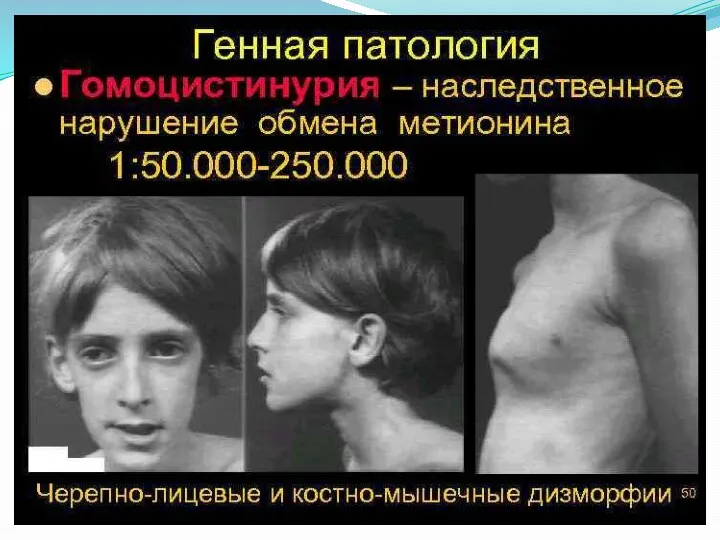
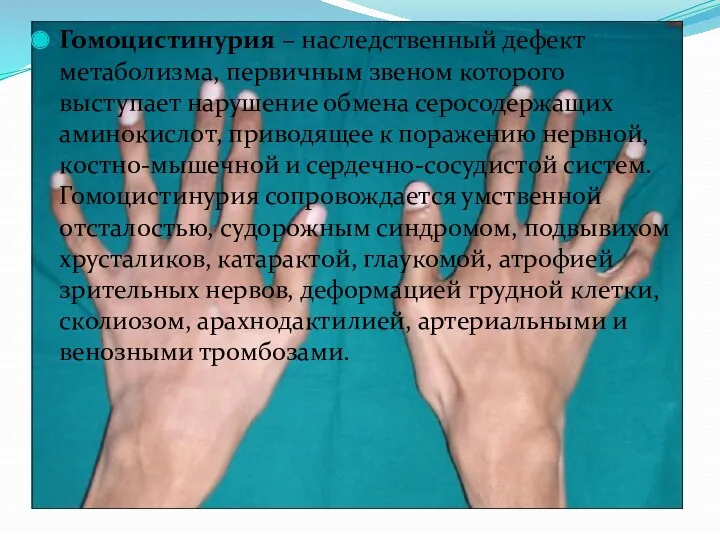
- 26. Гомоцистинурия – наследственный дефект метаболизма, первичным звеном которого выступает нарушение обмена серосодержащих аминокислот, приводящее к поражению
- 28. Симптомы гомоцистинурии Проявления гомоцистинурии нарастают постепенно. Дети рождаются без каких-либо специфических отклонений. В течение первого года
- 31. Поражение опорно-двигательного аппарата включает килевидную деформацию грудной клетки, арахнодактилию, кифосколиоз, остеопороз, искривление голеней, полую стопу или
- 35. Диагностика гомоцистинурии включает медико-генетическое консультирование, биохимическое исследование крови и мочи, офтальмологическое обследование, рентген-диагностику костной системы. Диагноз
- 36. Терапия гомоцистинурии проводится с учетом формы заболевания и включает диетическое питание, прием витаминов группы В. Лечебная
- 37. Цистинурия Цистинурия — это наследственное заболевание с нарушением белкового обмена, при котором не происходит всасывание в
- 38. Признаки болезни: интенсивные боли в поясничной области либо в боку, учащенное болезненное мочеиспускание, примесь крови в
- 40. Форма 1 тип - отсутствие транспорта цистина (незаменимая аминокислота для организма) и других аминокислот в кишечнике;
- 41. Диагностика Сбор анамнеза и жалоб заболевания: выраженный болевой синдром с характерной локализацией, беспокойное поведение больного, учащенное
- 42. Лечение цистинурии Лечение должно быть направлено на снижение количества незаменимой аминокислоты — цистина — в организме.
- 44. Алкаптонурия 1:250 тыс.-1:1 млн. людей в мире Алкаптонурия – генетически обусловленное нарушение метаболизма, характеризующееся врожденным дефицитом
- 46. Алкаптонурия характеризуется следующими основными симптомокомплексами: гомогентизиновой ацидурией, охронозом и артропатией. Эти признаки возникают в разное время:
- 48. Типичным признаком алкаптонурии служит уплотнение и серо-голубое окрашивание ушных раковин, пигментация склеры и конъюнктивы. Диффузное отложение
- 49. Диагностика алкаптонурии Чаще всего алкаптонурия диагностируется еще в раннем детском возрасте, однако в ряде случаев может
- 50. Лечение алкаптонурии Этиопатогенетическая терапия генетической алкаптонурии на сегодняшний день не разработана. Для предотвращения избыточного образования гомогентизиновой
- 52. Скачать презентацию

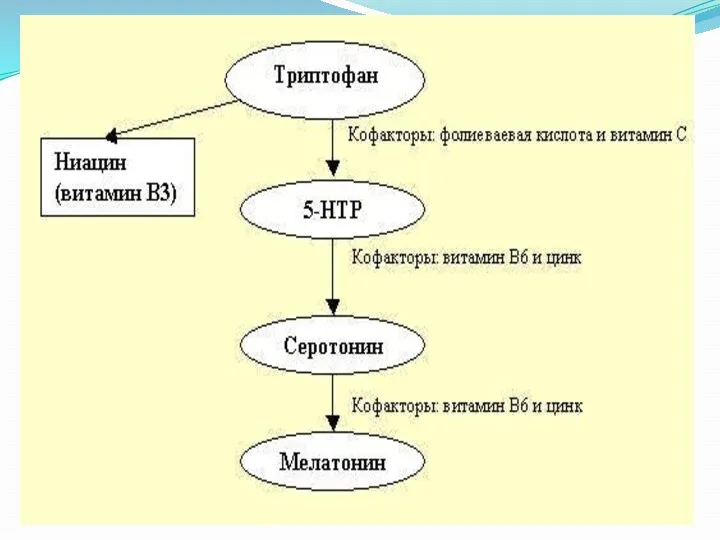
Триптофан является одной из 8-ми незаменимых для взрослого человека аминокислот. Относится к
Триптофан является одной из 8-ми незаменимых для взрослого человека аминокислот. Относится к

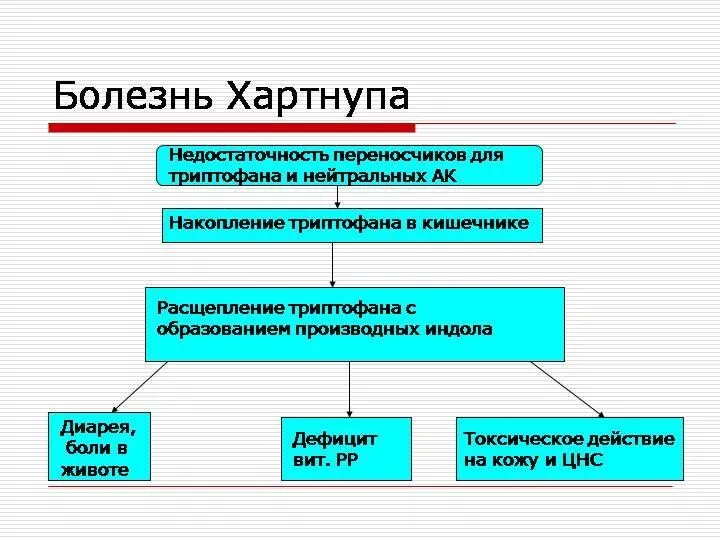
Болезнь Хартнупа
Болезнь Хартнупа — это редкое наследственное заболевание, в основе которого
Болезнь Хартнупа
Болезнь Хартнупа — это редкое наследственное заболевание, в основе которого

Этиопатогенез
При синдроме Хартнупа (имя больного, родители которого были двоюродными братом и сестрой) в
Этиопатогенез
При синдроме Хартнупа (имя больного, родители которого были двоюродными братом и сестрой) в

Причиной данного заболевания является мутация гена, который отвечает за обмен аминокислот,
Причиной данного заболевания является мутация гена, который отвечает за обмен аминокислот,

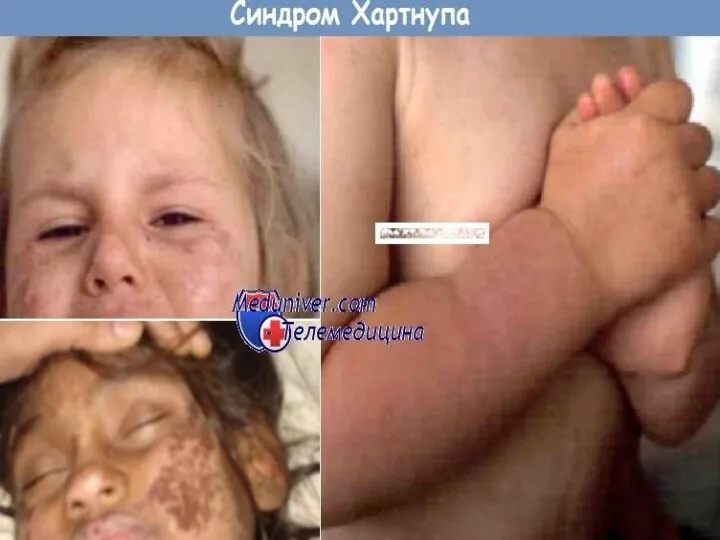
Кожа
Чаще поражению подвергаются воздействию солнечных лучей, а именно кожа
Кожа
Чаще поражению подвергаются воздействию солнечных лучей, а именно кожа
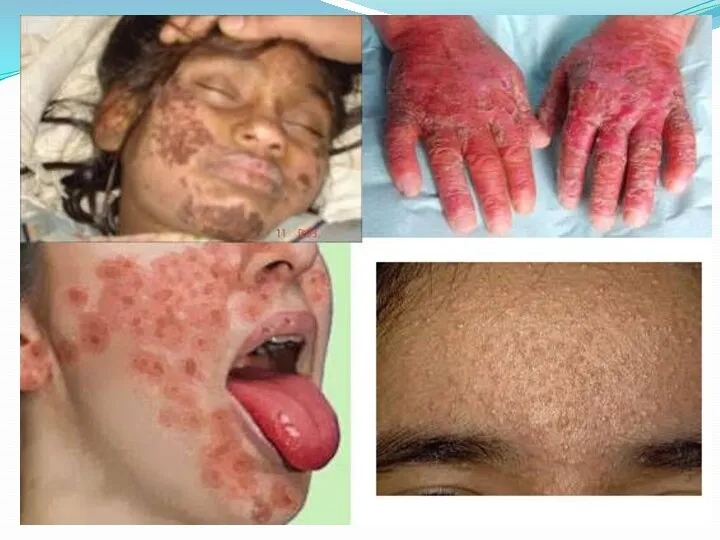
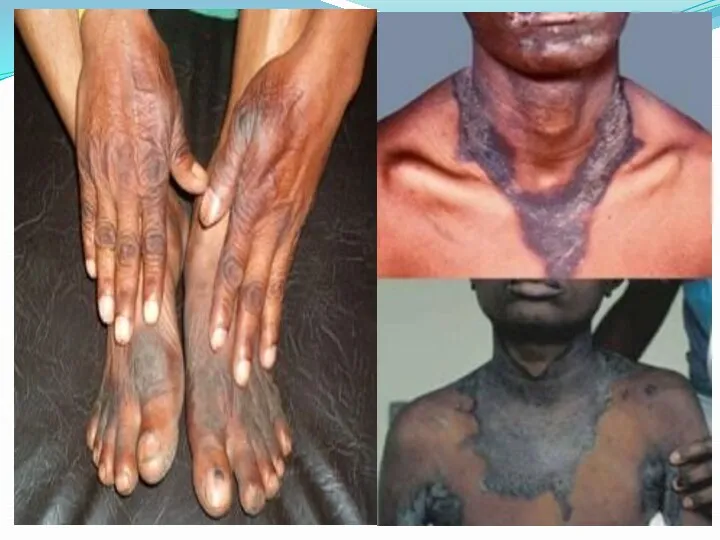
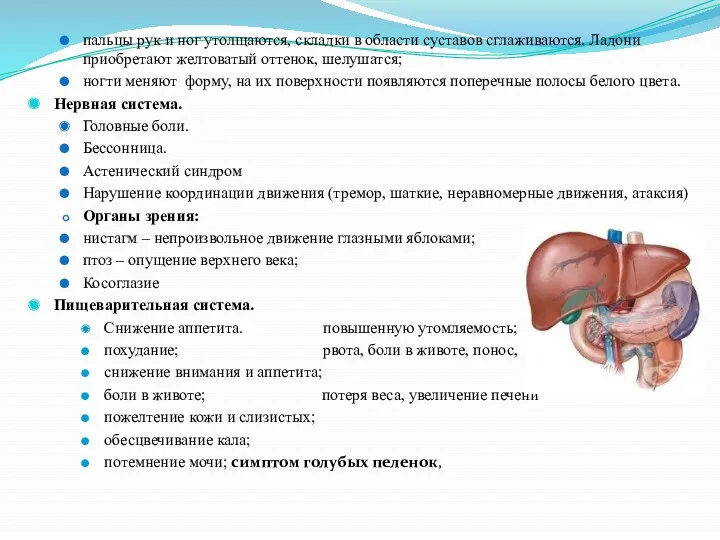
пальцы рук и ног утолщаются, складки в области суставов сглаживаются. Ладони
пальцы рук и ног утолщаются, складки в области суставов сглаживаются. Ладони

Факторы, способствующие развитию данного заболевания
плохое питание (недоедание; употребление продуктов, в которых
Факторы, способствующие развитию данного заболевания
плохое питание (недоедание; употребление продуктов, в которых

Диагностика
Общий осмотр — видны изменения:
Сбор анамнеза (у родственников больного выясняется наличие
Диагностика
Общий осмотр — видны изменения:
Сбор анамнеза (у родственников больного выясняется наличие

Лечение болезни Хартнупа
Больному назначают длительный прием витамина РР (никотиновой кислоты) и
Лечение болезни Хартнупа
Больному назначают длительный прием витамина РР (никотиновой кислоты) и

ЦИСТАТИОНИНУРИЯ
(цистатионин -f- греческий uron моча) — наследственная аномалия обмена серосодержащих аминокислот,
ЦИСТАТИОНИНУРИЯ
(цистатионин -f- греческий uron моча) — наследственная аномалия обмена серосодержащих аминокислот,
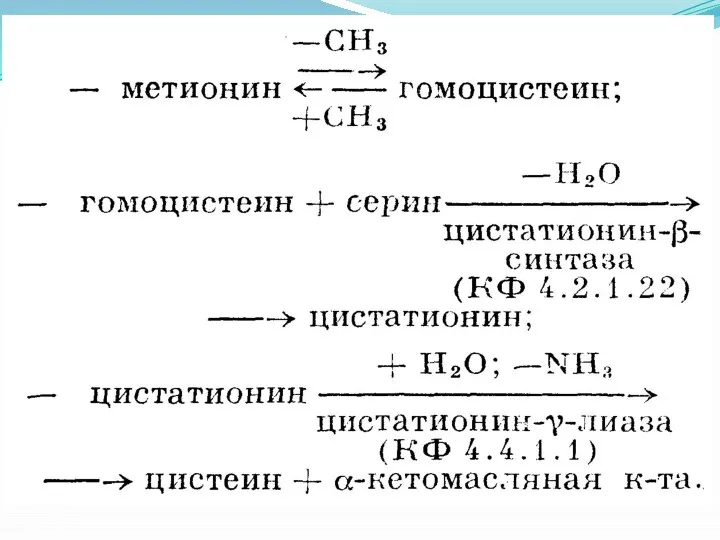

Клиническая картина характеризуется задержкой умственного развития ребенка. У некоторых больных с
Клиническая картина характеризуется задержкой умственного развития ребенка. У некоторых больных с

Гомоцистинурия
Это наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот,
Гомоцистинурия
Это наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот,
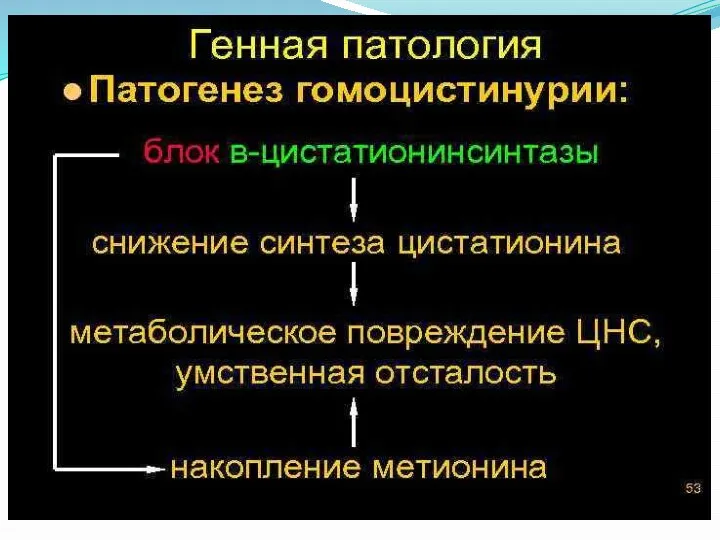


Установлено, что появление и увеличение концентрации гомоцистина в сыворотке крови способствует
Установлено, что появление и увеличение концентрации гомоцистина в сыворотке крови способствует
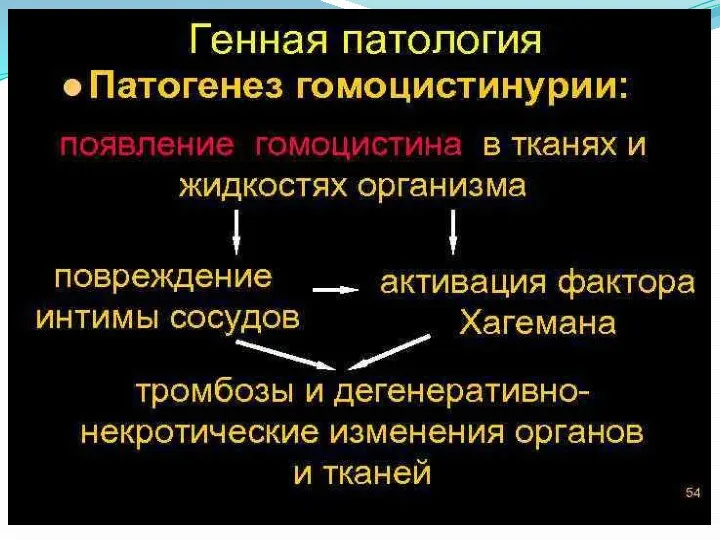
Гомоцистинурия – наследственный дефект метаболизма, первичным звеном которого выступает нарушение обмена серосодержащих
Гомоцистинурия – наследственный дефект метаболизма, первичным звеном которого выступает нарушение обмена серосодержащих

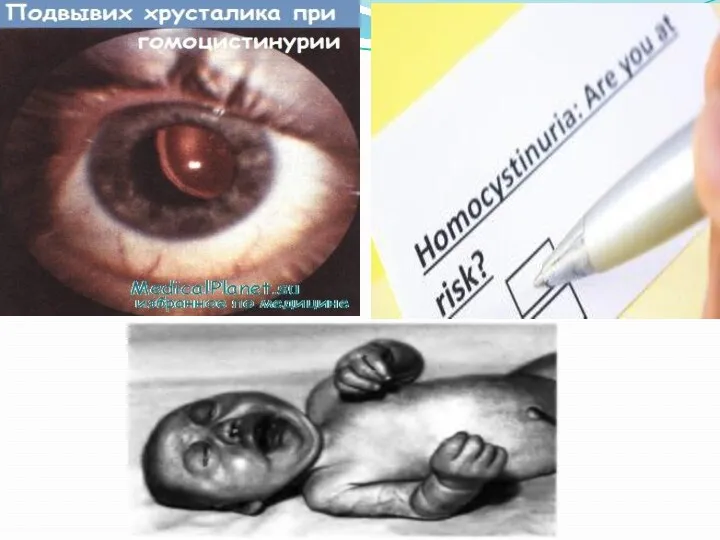
Симптомы гомоцистинурии
Проявления гомоцистинурии нарастают постепенно. Дети рождаются без каких-либо специфических отклонений.
Симптомы гомоцистинурии
Проявления гомоцистинурии нарастают постепенно. Дети рождаются без каких-либо специфических отклонений.


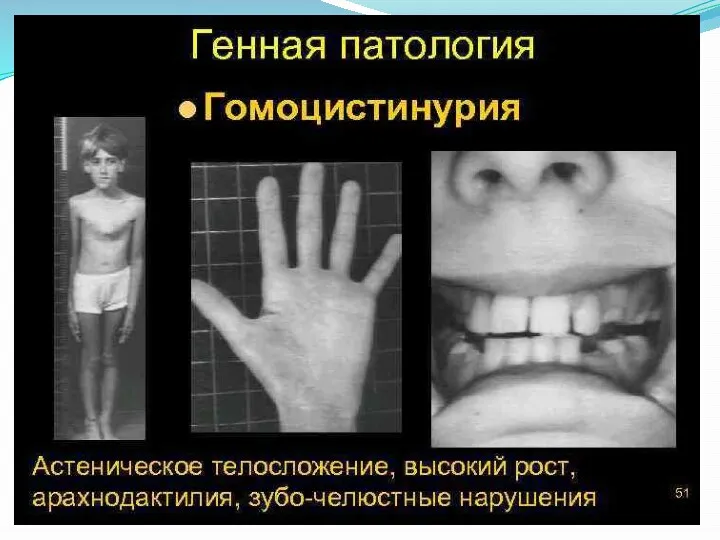
Поражение опорно-двигательного аппарата включает килевидную деформацию грудной клетки, арахнодактилию, кифосколиоз, остеопороз, искривление голеней, полую стопу или плоскостопие, готическое нёбо.
Поражение опорно-двигательного аппарата включает килевидную деформацию грудной клетки, арахнодактилию, кифосколиоз, остеопороз, искривление голеней, полую стопу или плоскостопие, готическое нёбо.
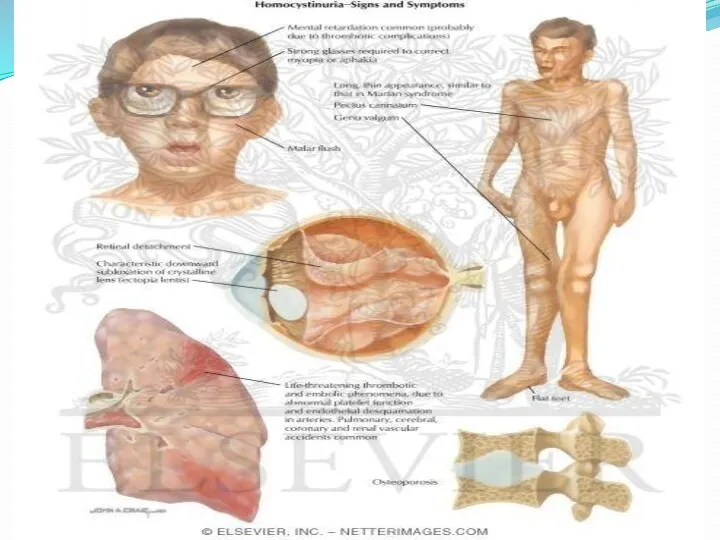
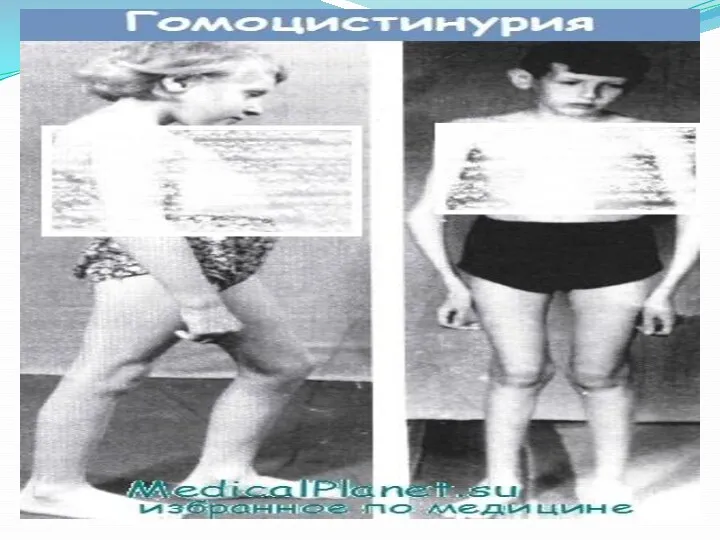

Диагностика гомоцистинурии включает медико-генетическое консультирование, биохимическое исследование крови и мочи, офтальмологическое
Диагностика гомоцистинурии включает медико-генетическое консультирование, биохимическое исследование крови и мочи, офтальмологическое

Терапия гомоцистинурии проводится с учетом формы заболевания и включает диетическое питание,
Терапия гомоцистинурии проводится с учетом формы заболевания и включает диетическое питание,

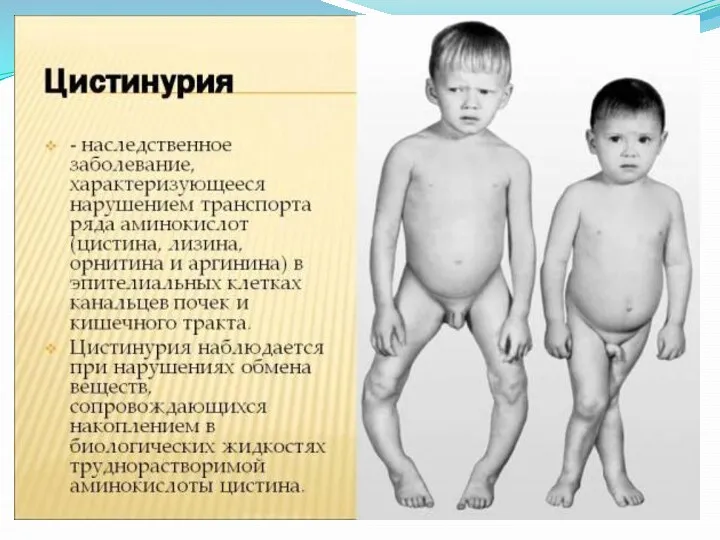
Цистинурия
Цистинурия — это наследственное заболевание с нарушением белкового обмена, при котором
Цистинурия
Цистинурия — это наследственное заболевание с нарушением белкового обмена, при котором
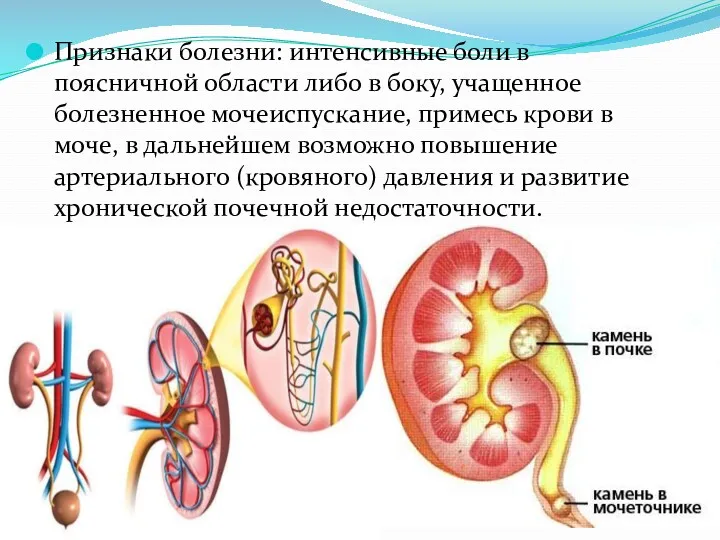
Признаки болезни: интенсивные боли в поясничной области либо в боку, учащенное
Признаки болезни: интенсивные боли в поясничной области либо в боку, учащенное

Форма
1 тип - отсутствие транспорта цистина (незаменимая аминокислота для организма)
Форма
1 тип - отсутствие транспорта цистина (незаменимая аминокислота для организма)

Диагностика
Сбор анамнеза и жалоб заболевания: выраженный болевой синдром с характерной локализацией,
Диагностика
Сбор анамнеза и жалоб заболевания: выраженный болевой синдром с характерной локализацией,

Лечение цистинурии
Лечение должно быть направлено на снижение количества незаменимой аминокислоты —
Лечение цистинурии
Лечение должно быть направлено на снижение количества незаменимой аминокислоты —


Алкаптонурия 1:250 тыс.-1:1 млн. людей в мире
Алкаптонурия – генетически обусловленное нарушение метаболизма,
Алкаптонурия 1:250 тыс.-1:1 млн. людей в мире
Алкаптонурия – генетически обусловленное нарушение метаболизма,

Алкаптонурия характеризуется следующими основными симптомокомплексами: гомогентизиновой ацидурией, охронозом и артропатией. Эти признаки
Алкаптонурия характеризуется следующими основными симптомокомплексами: гомогентизиновой ацидурией, охронозом и артропатией. Эти признаки
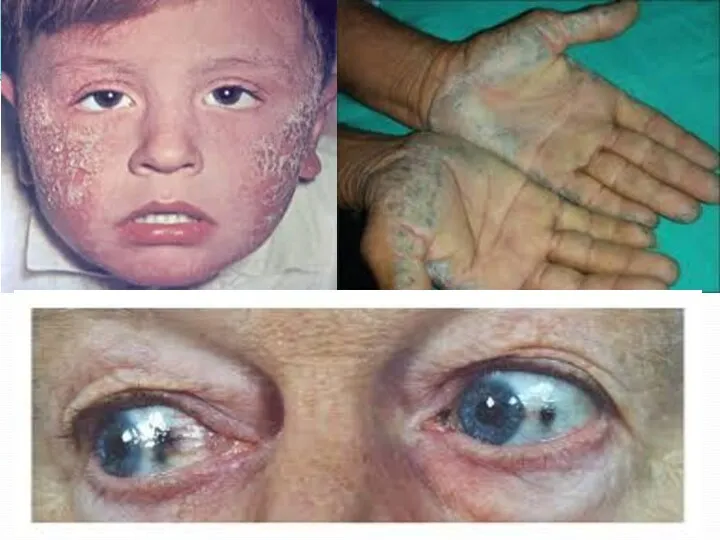
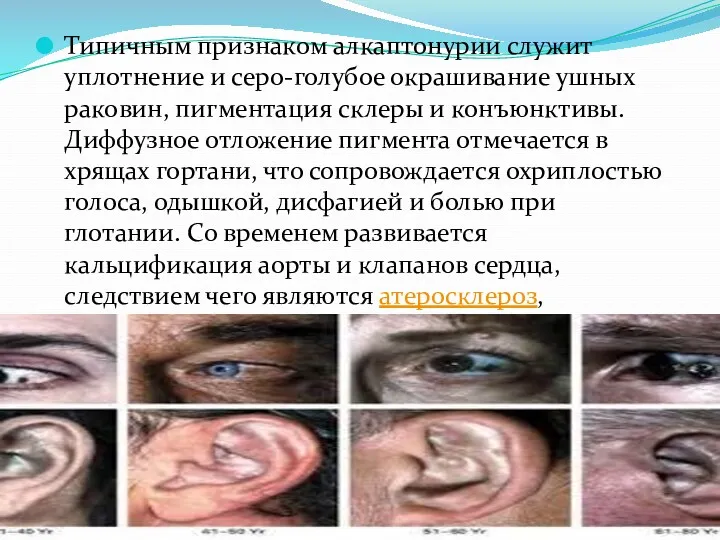
Типичным признаком алкаптонурии служит уплотнение и серо-голубое окрашивание ушных раковин, пигментация
Типичным признаком алкаптонурии служит уплотнение и серо-голубое окрашивание ушных раковин, пигментация

Диагностика алкаптонурии
Чаще всего алкаптонурия диагностируется еще в раннем детском возрасте, однако
Диагностика алкаптонурии
Чаще всего алкаптонурия диагностируется еще в раннем детском возрасте, однако

Лечение алкаптонурии
Этиопатогенетическая терапия генетической алкаптонурии на сегодняшний день не разработана. Для
Лечение алкаптонурии
Этиопатогенетическая терапия генетической алкаптонурии на сегодняшний день не разработана. Для
 Деятельность медицинской сестры в осуществлении мероприятий по профилактике миопии среди школьников
Деятельность медицинской сестры в осуществлении мероприятий по профилактике миопии среди школьников Мениски. Шов менисков или резекция? Что выбрать при повреждениях менисков коленного сустава?
Мениски. Шов менисков или резекция? Что выбрать при повреждениях менисков коленного сустава? Биожүйелердің электрөткізгіштігі
Биожүйелердің электрөткізгіштігі Узкий таз в акушерстве
Узкий таз в акушерстве Условные предложения. Виды условных предложений
Условные предложения. Виды условных предложений Грязелечение
Грязелечение Адресная доставка лекарственных нанопрепаратов в применении к моделям рака на доклиническом этапе исследований
Адресная доставка лекарственных нанопрепаратов в применении к моделям рака на доклиническом этапе исследований Гигиенические основы физического воспитания и закаливания детей и подростков
Гигиенические основы физического воспитания и закаливания детей и подростков Профессия врача. Одна из самых нужных и важных профессий
Профессия врача. Одна из самых нужных и важных профессий Самостоятельная работа интерна. Нарушения пищевого поведения
Самостоятельная работа интерна. Нарушения пищевого поведения Скульптурное моделирование лица
Скульптурное моделирование лица Перитонит, панкреатит, сепсис
Перитонит, панкреатит, сепсис Природно-очаговое заболевание чума
Природно-очаговое заболевание чума Pulp involvement
Pulp involvement ВИЧ-инфекция и СПИД
ВИЧ-инфекция и СПИД МРТ в диагностики ишемического инсульта. Сосудистая патология. Лекция 3
МРТ в диагностики ишемического инсульта. Сосудистая патология. Лекция 3 Сибирская язва. Возбудитель, эпидемиология, патогенез, диагностика, лечение, профилактика
Сибирская язва. Возбудитель, эпидемиология, патогенез, диагностика, лечение, профилактика Өкпе туберкулезі
Өкпе туберкулезі Почки. Анатомия почки. Нефрэктомия
Почки. Анатомия почки. Нефрэктомия Сахарный диабет у детей
Сахарный диабет у детей Тромбоэмолдық синдром
Тромбоэмолдық синдром Классификации психического дизонтогенеза
Классификации психического дизонтогенеза Медики - структура. Структура медицинского образования в Германии
Медики - структура. Структура медицинского образования в Германии Принципы рационального питания
Принципы рационального питания Сравнительный анализ методов хирургического лечения стрессового недержания мочи у женщин
Сравнительный анализ методов хирургического лечения стрессового недержания мочи у женщин Генетика негіздері
Генетика негіздері Склеродермия. Этиология склеродермии
Склеродермия. Этиология склеродермии Орнитоз (пситтакоз)
Орнитоз (пситтакоз)