- Аномалии зубов. Тема №4

Содержание
- 2. 1.Аномалии количества зубов. Наследственные нарушения формирования структуры зубов. 2. Генетические факторы агенезии зубов. 3. Наследственные заболевания
- 3. Множественные этиологические факторы способствуют нарушению гармоничного развития челюстно-лицевой области, что приводит к формированию аномалий количества зубов
- 4. К аномалиям количества зубов относят увеличение (гиперодонтия), уменьшение (гиподнонтия) или отсутствие зубов (адентия) по сравнению с
- 5. Генетические признаки могут быть обусловлены наследуемыми от родителей особенностями зубочелюстной системы - размерами и формой зу
- 6. Агенезия зубов (олигодентия, гиподентия, адентия) – это врожденное отсутствие одного или более молочных или постоянных зубов.
- 7. Семейная агенезия зубов наследуется по аутосомно-доминантному, аутосомно-рецессивному и X-сцепленному типу, но может и не проявлять четкого
- 8. ТРИХО-ОДОНТО-ОНИХИАЛЬНАЯ ДИСПЛАЗИЯ. Заболевание описано как новая форма эктодермальной дисплазии. Характерными признаками данного заболевания являются: гипоплазия эмали,
- 9. СИНДРОМ ХАЛЛЕРМАНА-ШТРАЙФА Для данного синдрома характерными признаками являются: частичная анодентия, наличие так называемых неонатальных зубов, малокклюзия,
- 10. СИНДРОМ ГАПО Синдром ГАПО - редкое аутосомно-рецессивное заболевание, которое вызывает серьезную задержку роста. ГАПО - это
- 11. РОТО-ЛИЦЕ-ПАЛЬЦЕВОЙ СИНДРОМ Синдром, характеризующийся расщеплением губы, дольчатым узловатым языком, широким корнем носа, гипоплазией тела нижней челюсти
- 12. ЛАКРИМО-АУРИКОЛО-ДЕНТО-ДИГИТАЛЬНЫЙ СИНДРОМ (ЛЕВИ-ХОЛЛИСТЕРА) Холлистер в 1973 г., Леви в 1969 г. описали заболевание, впоследствии названное их
- 13. НАСЛЕДСТВЕННАЯ ОСТЕОДИСТРОФИЯ АЛЬБРЕХТА. Это гетерогенное по своим проявлениям наследственное аутосомно-доминантное заболевание характеризуется низким ростом, ожирением, катарактой,
- 14. МУКОПОЛИСАХАРИДОЗЫ Мукополисахаридозы — это группа редких наследственных заболеваний соединительной ткани, связанных с нарушением обмена веществ. Они
- 15. ПАТОГЕНЕЗ МУКОПОЛИСАХАРИДОЗА Все формы мукополисахаридоза наследуются по аутосомно-рецессивному типу, т. е. мутировавший ген должен быть у
- 16. НЕЙРОСЕНСОРНАЯ ТУГОУХОСТЬ С ГИПОПЛАЗИЕЙ ЭМАЛИ И ДЕФЕКТАМИ НОГТЕЙ. Заболевание впервые описано в 1991 г. У 11-летнего
- 17. СТЕЙВА-ВИДЕМАННА СИНДРОМ Локализация гена LIFR, в области 5p13.1. Аутосомно-рецессивное заболевание, клиническими признаками которого являются: отставание в
- 18. СЕККЕЛЯ СИНДРОМ. Клинически гетерогенное аутосомно-рецессивное заболевание. Ген локализован в области 3q22-q24. Характеризуется отставанием в росте, умственной
- 19. ОКУЛО-ЦЕРЕБРО-РЕНАЛЬНЫЙ, СИНДРОМ ЛОУ Х-сцепленное заболевание, обусловлено мутацией в гене OCRL1. Ген локализован в области Xq26.1. Данное
- 20. СИНДРОМ МОРКИО Синдром Моркио – наследственная болезнь, которая обусловлена дефицитом в организме ферментов галактозамин-6-сульфат-сульфатазы или β-галактозидазы
- 21. БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ представляет собой группу редких наследственных заболеваний, характеризующихся нарушением межклеточных контактов в эпидермисе или дерме,
- 23. Скачать презентацию

1.Аномалии количества зубов. Наследственные нарушения формирования структуры зубов.
2. Генетические факторы агенезии
1.Аномалии количества зубов. Наследственные нарушения формирования структуры зубов.
2. Генетические факторы агенезии
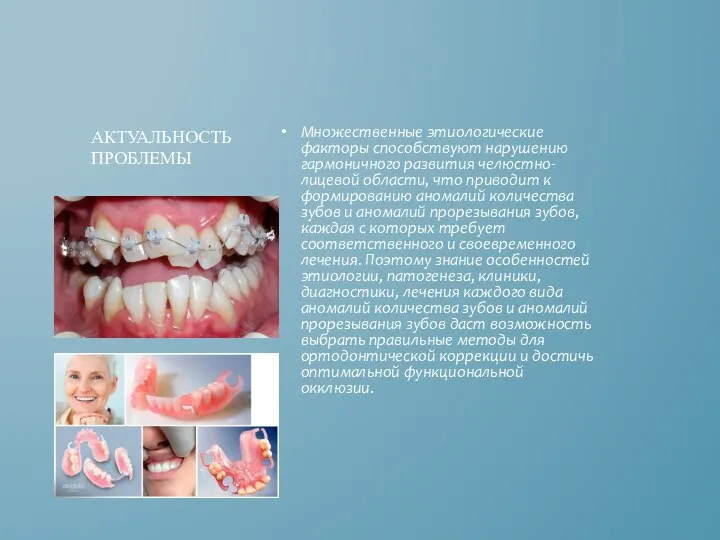
Множественные этиологические факторы способствуют нарушению гармоничного развития челюстно-лицевой области, что приводит
Множественные этиологические факторы способствуют нарушению гармоничного развития челюстно-лицевой области, что приводит
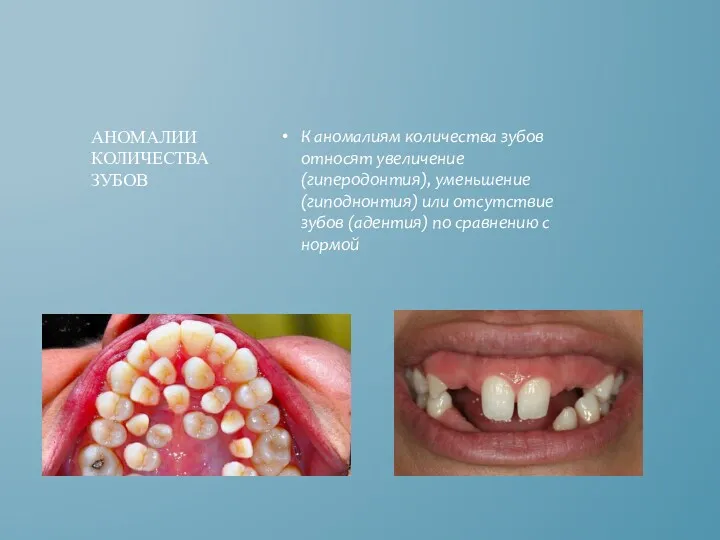
К аномалиям количества зубов относят увеличение (гиперодонтия), уменьшение (гиподнонтия) или отсутствие
К аномалиям количества зубов относят увеличение (гиперодонтия), уменьшение (гиподнонтия) или отсутствие

Генетические признаки могут быть обусловлены наследуемыми от родителей особенностями зубочелюстной системы
Генетические признаки могут быть обусловлены наследуемыми от родителей особенностями зубочелюстной системы
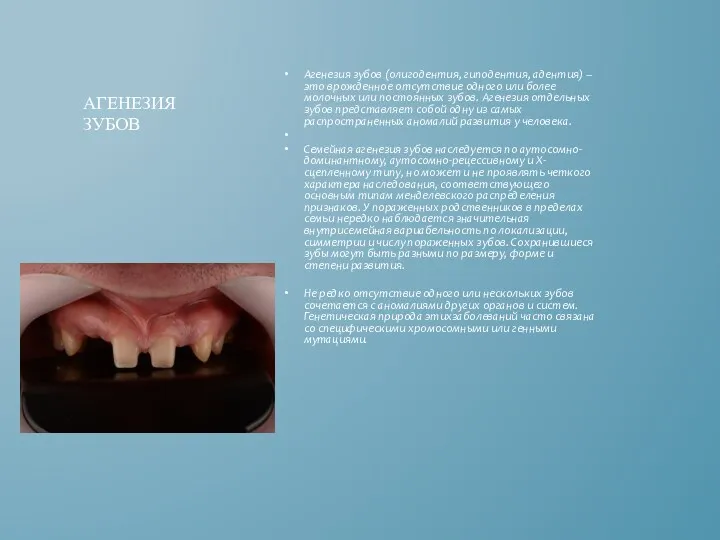
Агенезия зубов (олигодентия, гиподентия, адентия) – это врожденное отсутствие одного или
Агенезия зубов (олигодентия, гиподентия, адентия) – это врожденное отсутствие одного или

Семейная агенезия зубов наследуется по аутосомно-доминантному, аутосомно-рецессивному и X-сцепленному типу, но
Семейная агенезия зубов наследуется по аутосомно-доминантному, аутосомно-рецессивному и X-сцепленному типу, но
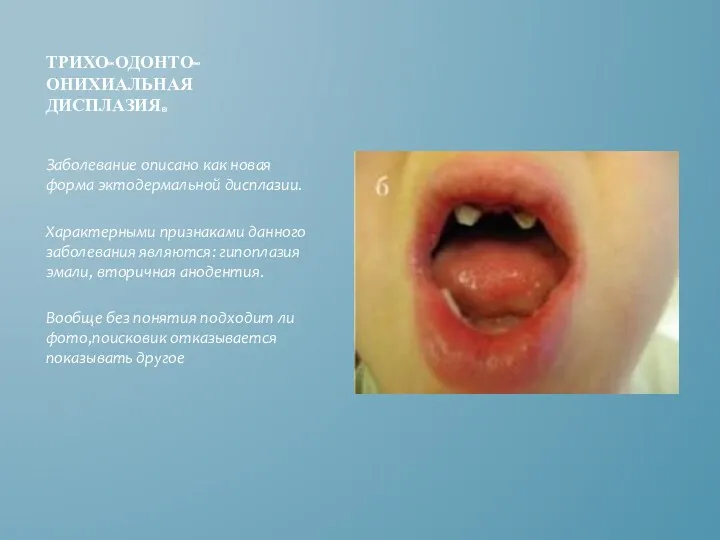
ТРИХО-ОДОНТО-ОНИХИАЛЬНАЯ ДИСПЛАЗИЯ.
Заболевание описано как новая форма эктодермальной дисплазии.
Характерными признаками данного
ТРИХО-ОДОНТО-ОНИХИАЛЬНАЯ ДИСПЛАЗИЯ.
Заболевание описано как новая форма эктодермальной дисплазии.
Характерными признаками данного
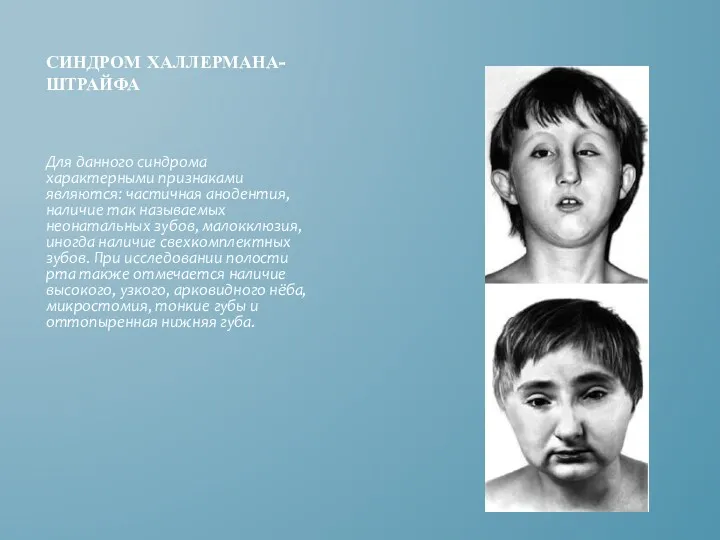
СИНДРОМ ХАЛЛЕРМАНА-ШТРАЙФА
Для данного синдрома характерными признаками являются: частичная анодентия, наличие
СИНДРОМ ХАЛЛЕРМАНА-ШТРАЙФА
Для данного синдрома характерными признаками являются: частичная анодентия, наличие

СИНДРОМ ГАПО
Синдром ГАПО - редкое аутосомно-рецессивное заболевание, которое вызывает серьезную задержку
СИНДРОМ ГАПО
Синдром ГАПО - редкое аутосомно-рецессивное заболевание, которое вызывает серьезную задержку
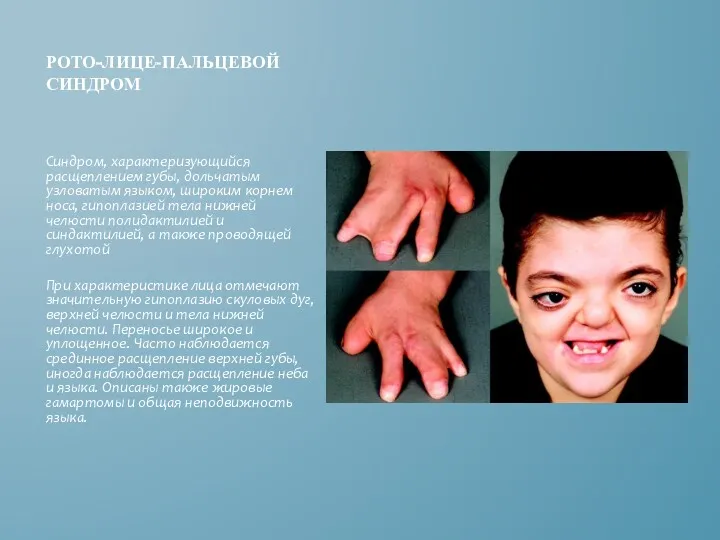
РОТО-ЛИЦЕ-ПАЛЬЦЕВОЙ СИНДРОМ
Синдром, характеризующийся расщеплением губы, дольчатым узловатым языком, широким корнем носа,
РОТО-ЛИЦЕ-ПАЛЬЦЕВОЙ СИНДРОМ
Синдром, характеризующийся расщеплением губы, дольчатым узловатым языком, широким корнем носа,

ЛАКРИМО-АУРИКОЛО-ДЕНТО-ДИГИТАЛЬНЫЙ СИНДРОМ (ЛЕВИ-ХОЛЛИСТЕРА)
Холлистер в 1973 г., Леви в 1969 г. описали
ЛАКРИМО-АУРИКОЛО-ДЕНТО-ДИГИТАЛЬНЫЙ СИНДРОМ (ЛЕВИ-ХОЛЛИСТЕРА)
Холлистер в 1973 г., Леви в 1969 г. описали

НАСЛЕДСТВЕННАЯ ОСТЕОДИСТРОФИЯ АЛЬБРЕХТА.
Это гетерогенное по своим проявлениям наследственное аутосомно-доминантное заболевание характеризуется
НАСЛЕДСТВЕННАЯ ОСТЕОДИСТРОФИЯ АЛЬБРЕХТА.
Это гетерогенное по своим проявлениям наследственное аутосомно-доминантное заболевание характеризуется

МУКОПОЛИСАХАРИДОЗЫ
Мукополисахаридозы — это группа редких наследственных заболеваний соединительной ткани, связанных с
МУКОПОЛИСАХАРИДОЗЫ
Мукополисахаридозы — это группа редких наследственных заболеваний соединительной ткани, связанных с
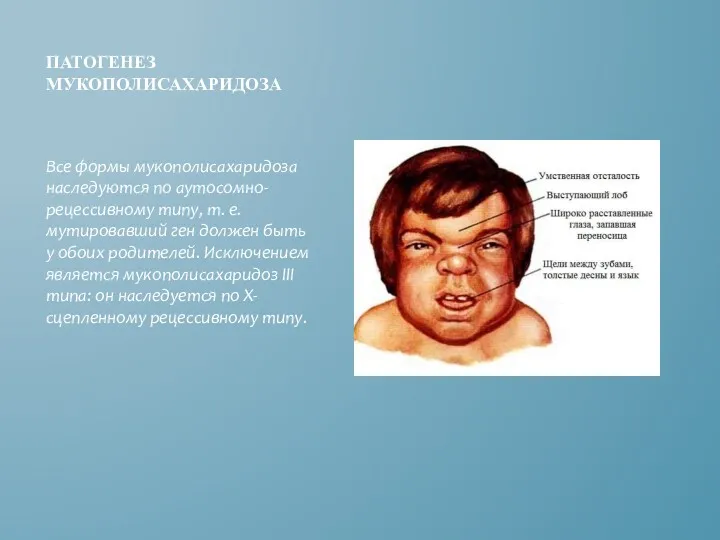
ПАТОГЕНЕЗ МУКОПОЛИСАХАРИДОЗА
Все формы мукополисахаридоза наследуются по аутосомно-рецессивному типу, т. е. мутировавший
ПАТОГЕНЕЗ МУКОПОЛИСАХАРИДОЗА
Все формы мукополисахаридоза наследуются по аутосомно-рецессивному типу, т. е. мутировавший
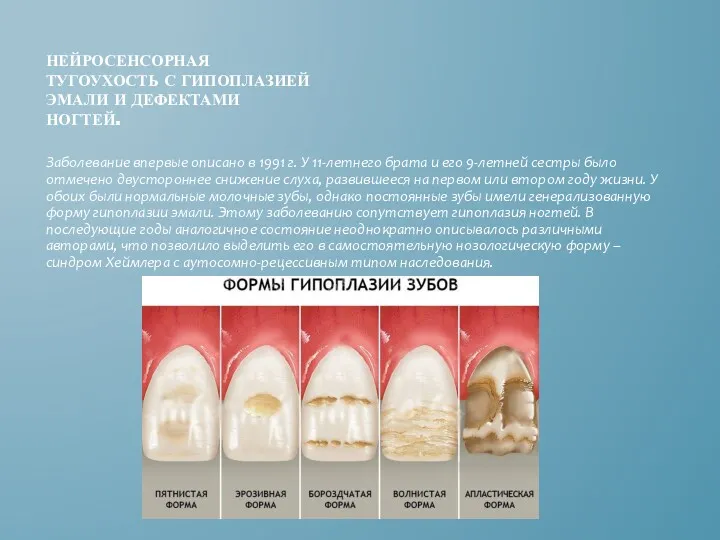
НЕЙРОСЕНСОРНАЯ ТУГОУХОСТЬ С ГИПОПЛАЗИЕЙ ЭМАЛИ И ДЕФЕКТАМИ НОГТЕЙ.
Заболевание впервые описано в
НЕЙРОСЕНСОРНАЯ ТУГОУХОСТЬ С ГИПОПЛАЗИЕЙ ЭМАЛИ И ДЕФЕКТАМИ НОГТЕЙ.
Заболевание впервые описано в
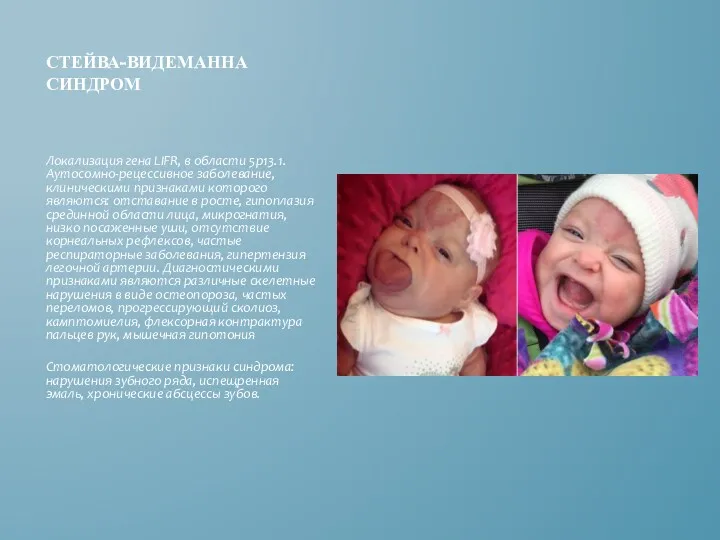
СТЕЙВА-ВИДЕМАННА СИНДРОМ
Локализация гена LIFR, в области 5p13.1. Аутосомно-рецессивное заболевание, клиническими признаками
СТЕЙВА-ВИДЕМАННА СИНДРОМ
Локализация гена LIFR, в области 5p13.1. Аутосомно-рецессивное заболевание, клиническими признаками
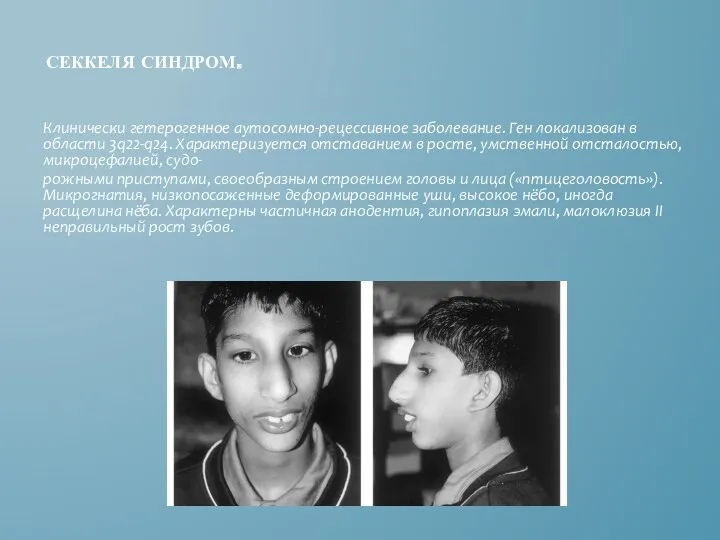
СЕККЕЛЯ СИНДРОМ.
Клинически гетерогенное аутосомно-рецессивное заболевание. Ген локализован в области 3q22-q24. Характеризуется
СЕККЕЛЯ СИНДРОМ.
Клинически гетерогенное аутосомно-рецессивное заболевание. Ген локализован в области 3q22-q24. Характеризуется

ОКУЛО-ЦЕРЕБРО-РЕНАЛЬНЫЙ, СИНДРОМ ЛОУ
Х-сцепленное заболевание, обусловлено мутацией в гене OCRL1. Ген локализован
ОКУЛО-ЦЕРЕБРО-РЕНАЛЬНЫЙ, СИНДРОМ ЛОУ
Х-сцепленное заболевание, обусловлено мутацией в гене OCRL1. Ген локализован
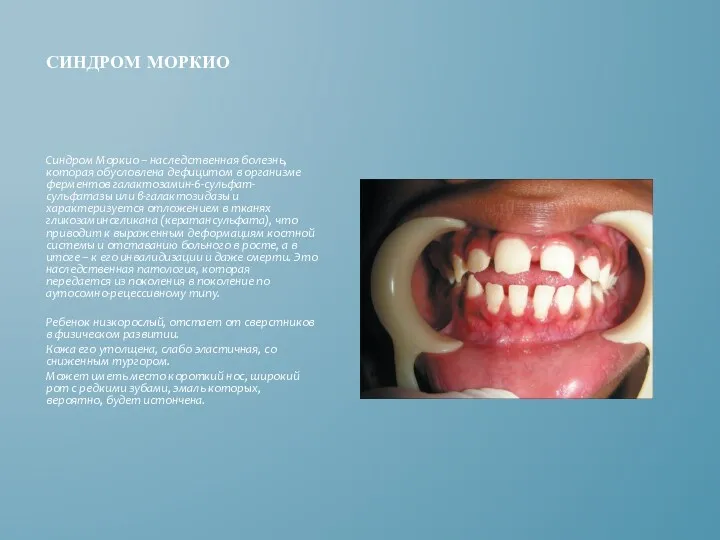
СИНДРОМ МОРКИО
Синдром Моркио – наследственная болезнь, которая обусловлена дефицитом в организме
СИНДРОМ МОРКИО
Синдром Моркио – наследственная болезнь, которая обусловлена дефицитом в организме

БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ
представляет собой группу редких наследственных заболеваний, характеризующихся нарушением межклеточных контактов
БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ
представляет собой группу редких наследственных заболеваний, характеризующихся нарушением межклеточных контактов
 Введение в психофармакологию. Средства для общей и местной анестезии
Введение в психофармакологию. Средства для общей и местной анестезии Общая физиология ЦНС
Общая физиология ЦНС Принципы, методы гемотрансфузий. Компоненты и препараты крови
Принципы, методы гемотрансфузий. Компоненты и препараты крови Дәрігер кәсіби имиджі және кәсіби бейімділігі
Дәрігер кәсіби имиджі және кәсіби бейімділігі Недоношенные дети: патология
Недоношенные дети: патология Деменции первичные и вторичные,
Деменции первичные и вторичные, Лечебная физкультура при гломерулонефрите
Лечебная физкультура при гломерулонефрите Жаңа туған нәрестенің физиологиясы және патологиясы
Жаңа туған нәрестенің физиологиясы және патологиясы Лечение хронической ишемической болезни сердца
Лечение хронической ишемической болезни сердца Артериальная гипертензия у детей и подростков (диагностика, лечение, профилактика)
Артериальная гипертензия у детей и подростков (диагностика, лечение, профилактика) Аномалії розвитку плідного яйця
Аномалії розвитку плідного яйця Острый панкреатит
Острый панкреатит Гениталды эндометриоз
Гениталды эндометриоз Мышцы нижней конечности. Мышцы тазового пояса. Мышцы свободной нижней конечности
Мышцы нижней конечности. Мышцы тазового пояса. Мышцы свободной нижней конечности Классификация дистонии
Классификация дистонии Первичные и вторичные гломерулопатии
Первичные и вторичные гломерулопатии Оценка качеств клинических руководств с помощью системы agree
Оценка качеств клинических руководств с помощью системы agree Радиобиологиялық әсерлер
Радиобиологиялық әсерлер How does smoking kill?
How does smoking kill? Лабораторная диагностика. Дополнительные методы обследования
Лабораторная диагностика. Дополнительные методы обследования Відмороження. Визначення поняття “відмороження”
Відмороження. Визначення поняття “відмороження” Шеміршек тіні
Шеміршек тіні Естественные защитные силы организма
Естественные защитные силы организма Фармацевт - провизор
Фармацевт - провизор Тактика ведения пациентов с аутоиммунной тромбоцитопенией
Тактика ведения пациентов с аутоиммунной тромбоцитопенией СТЖБ-ң анатомиясы, физиологиясы; СТЖБ-ң қызметінің бұзылысы; Артикулатемпералды синдром
СТЖБ-ң анатомиясы, физиологиясы; СТЖБ-ң қызметінің бұзылысы; Артикулатемпералды синдром ЛФК при нарушениях осанки детей младшего школьного возраста
ЛФК при нарушениях осанки детей младшего школьного возраста Руководство WSAVA по распознаванию, оценке и лечению боли
Руководство WSAVA по распознаванию, оценке и лечению боли