- Болезни аминокислотного обмена

Содержание
- 2. Фенилкетонурия (ФКУ) (E70.1) (12q24.1 РАН, PKU1 или - в случае дефицита дигидроптеридинредуктазы - 4q15.1) Частота 1:10
- 3. Наследственные болезни, связанные с нарушением липидного обмена 1. Болезни накопления (тезаурисмозы) - внутриклеточные липидозы, при которых
- 4. Болезнь Гоше .
- 7. Болезнь Гоше. Лечение в 1991 году в США появился первый медицинский препарат Аглюцераза, позволяющий лечить болезнь
- 9. Мукополисахаридоз, тип I (недостаточность фермента лизосомной a-L-идуронидазы, синдромы Гурлер, Гурлер-Шейе и Шейе) - аутосомно-рецессивное заболевание, возникающее
- 10. Патогенез мукополисахаридоза I типа Фермент a-L-идуронидаза участвует в метаболизме двух гликозаминогликанов - дерматан-сульфата и гепарансульфата. Поскольку
- 11. Мукополисахаридоз, тип IH (синдром Гурлер) клинические признаки заболевания появляются на первом году жизни, с пиком манифестации
- 13. Мукополисахаридоз, тип I-H/S (синдром Гурлер-Шейе) Мукополисахаридоз, тип I-H/S (синдром Гурлер-Шейе) Клинический фенотип синдрома Гурлер-Шейе занимает промежуточное
- 14. Мукополисахаридоз, тип IS (синдром Шейе) Мукополисахаридоз, тип IS (синдром Шейе) В первоначальной классификации мукополисахаридозов, до открытия
- 16. Скачать презентацию

Фенилкетонурия (ФКУ) (E70.1)
(12q24.1 РАН, PKU1 или - в случае дефицита
Фенилкетонурия (ФКУ) (E70.1)
(12q24.1 РАН, PKU1 или - в случае дефицита

Наследственные болезни, связанные с нарушением липидного обмена
1. Болезни накопления (тезаурисмозы) -
Наследственные болезни, связанные с нарушением липидного обмена
1. Болезни накопления (тезаурисмозы) -

Болезнь Гоше
.
Болезнь Гоше
.
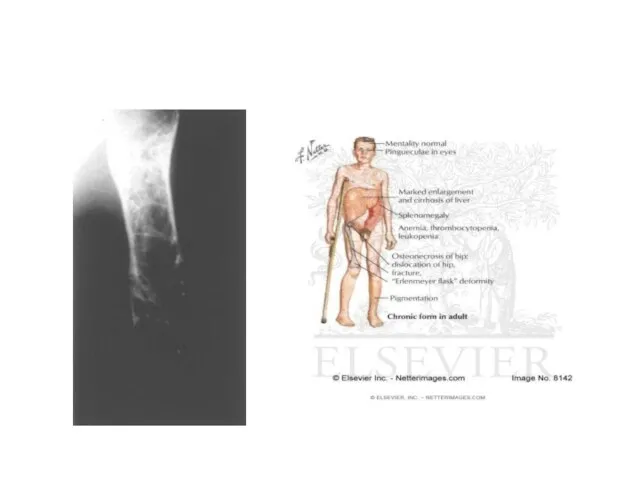


Болезнь Гоше. Лечение
в 1991 году в США появился первый медицинский препарат
Болезнь Гоше. Лечение
в 1991 году в США появился первый медицинский препарат


Мукополисахаридоз, тип I (недостаточность фермента лизосомной a-L-идуронидазы, синдромы Гурлер, Гурлер-Шейе и
Мукополисахаридоз, тип I (недостаточность фермента лизосомной a-L-идуронидазы, синдромы Гурлер, Гурлер-Шейе и

Патогенез мукополисахаридоза I типа
Фермент a-L-идуронидаза участвует в метаболизме двух гликозаминогликанов -
Патогенез мукополисахаридоза I типа
Фермент a-L-идуронидаза участвует в метаболизме двух гликозаминогликанов -

Мукополисахаридоз, тип IH (синдром Гурлер)
клинические признаки заболевания появляются на первом
Мукополисахаридоз, тип IH (синдром Гурлер)
клинические признаки заболевания появляются на первом
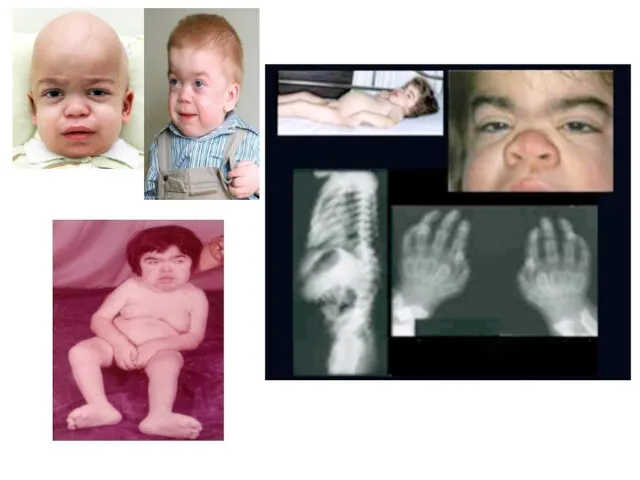

Мукополисахаридоз, тип I-H/S (синдром Гурлер-Шейе)
Мукополисахаридоз, тип I-H/S (синдром Гурлер-Шейе) Клинический фенотип
Мукополисахаридоз, тип I-H/S (синдром Гурлер-Шейе)
Мукополисахаридоз, тип I-H/S (синдром Гурлер-Шейе) Клинический фенотип

Мукополисахаридоз, тип IS (синдром Шейе)
Мукополисахаридоз, тип IS (синдром Шейе)
В первоначальной классификации
Мукополисахаридоз, тип IS (синдром Шейе)
Мукополисахаридоз, тип IS (синдром Шейе)
В первоначальной классификации
 Lazer kosmetologiya
Lazer kosmetologiya Массаж как неспецифическое средство ЛФК
Массаж как неспецифическое средство ЛФК Артериялық қысым
Артериялық қысым Организация ПМСП сельскому населению
Организация ПМСП сельскому населению Өндірістік токсикология негіздері. (Тақырып 8)
Өндірістік токсикология негіздері. (Тақырып 8) Ағзаларды клондау
Ағзаларды клондау Синдром приобретенного иммунного дефицита (СПИД) и ВИЧ
Синдром приобретенного иммунного дефицита (СПИД) и ВИЧ Ауыз қуысы кілігейлі шырышты қабатының, ерін ауруларының сырқатымен келген науқастырды тексеру әдістері
Ауыз қуысы кілігейлі шырышты қабатының, ерін ауруларының сырқатымен келген науқастырды тексеру әдістері Телекоммуникационные системы в медицине (ч.2)
Телекоммуникационные системы в медицине (ч.2) Нарушения периферического кровообращения
Нарушения периферического кровообращения Дислалия и дизартрия
Дислалия и дизартрия Флавоноиды. Лекарственные растения и лекарственное растительное сырье, содержащие флавоноиды
Флавоноиды. Лекарственные растения и лекарственное растительное сырье, содержащие флавоноиды Регуляция иммунного ответа. Гормональная регуляция иммунных реакций. Система цитокинов. Интерфероны. Факторы роста
Регуляция иммунного ответа. Гормональная регуляция иммунных реакций. Система цитокинов. Интерфероны. Факторы роста Рак шейки и тела матки
Рак шейки и тела матки Электрокардиограмма при гипертрофиях предсердий и желудочков. Электрокардиограмма при ишемической болезни сердца
Электрокардиограмма при гипертрофиях предсердий и желудочков. Электрокардиограмма при ишемической болезни сердца Особенности оказания неотложной медицинской помощи на догоспитальном этапе при черепно-мозговых травмах
Особенности оказания неотложной медицинской помощи на догоспитальном этапе при черепно-мозговых травмах Современные биохимические и иммунологические методы исследования в медицине
Современные биохимические и иммунологические методы исследования в медицине Балалардағы рефлюкс нефропатиялар. БМСККО жағдайында жүргізу әрекеті мен динамикалық бақылау
Балалардағы рефлюкс нефропатиялар. БМСККО жағдайында жүргізу әрекеті мен динамикалық бақылау Плацентарная недостаточность
Плацентарная недостаточность ЛФК при хирургических вмешательствах
ЛФК при хирургических вмешательствах Принципы, задачи, организация медицинской службы для оказания помощи населению в ЧС мирного и военного времени
Принципы, задачи, организация медицинской службы для оказания помощи населению в ЧС мирного и военного времени Оказание первой помощи в школьных условиях
Оказание первой помощи в школьных условиях Процессы приспособления (адаптации) и компенсации организма
Процессы приспособления (адаптации) и компенсации организма Первая помощь при травме
Первая помощь при травме Основные принципы химиотерапии
Основные принципы химиотерапии Практические проблемы проведения профилактического медицинского осмотра
Практические проблемы проведения профилактического медицинского осмотра Туляремия – острое природно-очаговое заболевание с группы бактериальных зоонозов
Туляремия – острое природно-очаговое заболевание с группы бактериальных зоонозов Осторожно: клещ
Осторожно: клещ