- Фенилкетонурия. Клиника

Содержание
- 2. Введение Актуальность темы Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина, и является наиболее распространенной
- 3. Этиология Мутация гена (т.е. его изменение), произошедшая по тем или иным причинам в области локализации 12
- 4. Классификация фенилкетонурий Фенилкетонурия I Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена, локализующегося в длинном плече 12
- 5. Фенилкетонурия I Дети с фенилкетонурией рождаются без каких-либо признаков болезни. Однако уже на втором месяце можно
- 6. В клинической картине ФКУ II преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, сухожильная гиперрефлексия, мышечная
- 7. Материнская фенилкетонурия Заболевание развивается у потомков женщин, страдающих ФКУ и не получающих диету в зрелом возрасте.
- 8. Диагностика. Диагноз основывается на совокупности генеалогических данных, результатов клинического и биохимического обследования: -клинические симптомы -возможный родственный
- 9. Диагностика Флуориметрия Определение концентрации фенилаланина при облучении ультрафиолетом. Позволяет обнаружить микродозы вещества. Тест Гатри Несколько капель
- 10. Проба Феллинга Это метод обнаружения фенилпировиноградной кислоты в моче К моче ребёнка добавляют уксусную кислоту и
- 11. Поиск мутантного гена Проводится прямой поиск мутантного гена с помощью синтетических олигонуклеотидных зондов. Помимо этого возможно
- 12. Лечение Диетотерапия Единственным лечением, способным предотвратить развитие слабоумия или уменьшить его степень, является диета, исключающая поступление
- 14. Допустимое суточное количество фенилаланина для больных ФКУ детей различных возрастных групп.
- 15. Лечение Медикаментозное лечение При стандартном, медикаментозном лечении фенилкетонурии, используют препараты ноотропной группы, лекарства, содержащие сбалансированное количество
- 16. Дополнительная терапия. -Массаж и лечебная физкультура. -Интеллектуальная реабилитация, в том числе с использованием современных компьютерных технологий:
- 17. Сестринский уход за детьми больными фенилкетонурией. Медицинская сестра обеспечивает: - соблюдение ребёнком диеты; - проветривание, кварцевание,
- 19. Скачать презентацию

Введение
Актуальность темы
Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина, и
Введение
Актуальность темы
Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина, и
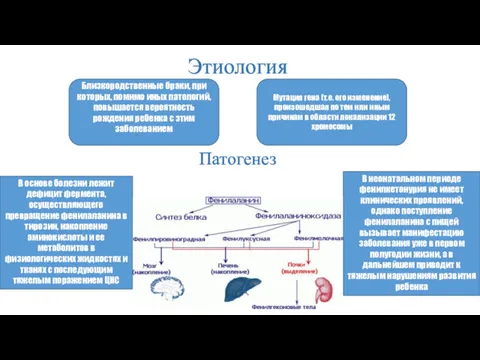
Этиология
Мутация гена (т.е. его изменение), произошедшая по тем или иным причинам
Этиология
Мутация гена (т.е. его изменение), произошедшая по тем или иным причинам

Классификация фенилкетонурий
Фенилкетонурия I
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена,
локализующегося в длинном плече
Классификация фенилкетонурий
Фенилкетонурия I
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена,
локализующегося в длинном плече

Фенилкетонурия I
Дети с фенилкетонурией рождаются без каких-либо признаков болезни.
Однако уже на втором
Фенилкетонурия I
Дети с фенилкетонурией рождаются без каких-либо признаков болезни.
Однако уже на втором
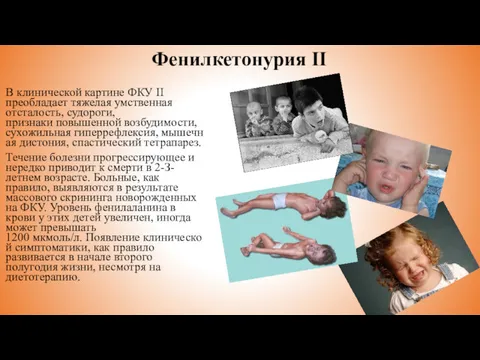
В клинической картине ФКУ II преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, сухожильная гиперрефлексия, мышечная дистония, спастический тетрапарез.
Течение
В клинической картине ФКУ II преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, сухожильная гиперрефлексия, мышечная дистония, спастический тетрапарез.
Течение
Материнская фенилкетонурия
Заболевание развивается у потомков женщин, страдающих ФКУ и не получающих
Материнская фенилкетонурия
Заболевание развивается у потомков женщин, страдающих ФКУ и не получающих
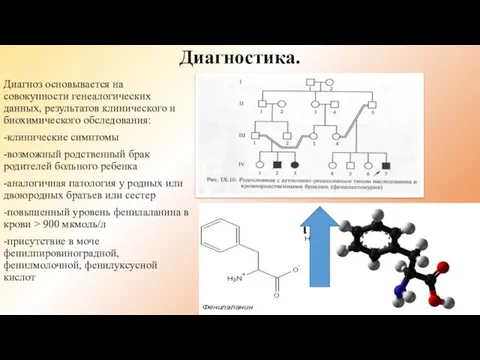
Диагностика.
Диагноз основывается на совокупности генеалогических данных, результатов клинического и биохимического обследования:
-клинические
Диагностика.
Диагноз основывается на совокупности генеалогических данных, результатов клинического и биохимического обследования:
-клинические

Диагностика
Флуориметрия
Определение концентрации фенилаланина при облучении ультрафиолетом. Позволяет обнаружить микродозы вещества.
Тест
Диагностика
Флуориметрия
Определение концентрации фенилаланина при облучении ультрафиолетом. Позволяет обнаружить микродозы вещества.
Тест

Проба Феллинга
Это метод обнаружения фенилпировиноградной кислоты в моче
К моче ребёнка
Проба Феллинга
Это метод обнаружения фенилпировиноградной кислоты в моче К моче ребёнка
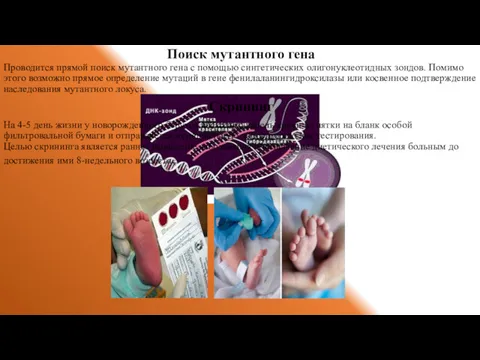
Поиск мутантного гена
Проводится прямой поиск мутантного гена с помощью синтетических олигонуклеотидных
Поиск мутантного гена
Проводится прямой поиск мутантного гена с помощью синтетических олигонуклеотидных

Лечение
Диетотерапия
Единственным лечением, способным предотвратить развитие слабоумия или уменьшить его степень,
Лечение
Диетотерапия Единственным лечением, способным предотвратить развитие слабоумия или уменьшить его степень,

Допустимое суточное количество фенилаланина для больных ФКУ детей различных возрастных групп.
Допустимое суточное количество фенилаланина для больных ФКУ детей различных возрастных групп.

Лечение
Медикаментозное лечение
При стандартном, медикаментозном лечении фенилкетонурии, используют препараты ноотропной группы,
Лечение
Медикаментозное лечение
При стандартном, медикаментозном лечении фенилкетонурии, используют препараты ноотропной группы,

Дополнительная терапия.
-Массаж и лечебная физкультура.
-Интеллектуальная реабилитация, в том числе с
Дополнительная терапия.
-Массаж и лечебная физкультура.
-Интеллектуальная реабилитация, в том числе с

Сестринский уход за детьми больными фенилкетонурией.
Медицинская сестра обеспечивает:
- соблюдение ребёнком диеты;
-
Сестринский уход за детьми больными фенилкетонурией.
Медицинская сестра обеспечивает:
- соблюдение ребёнком диеты;
-
 Фізична терапія при пульпозних форамінальних кілах поперекового відділу хребта у чоловіків віком 30-50 років
Фізична терапія при пульпозних форамінальних кілах поперекового відділу хребта у чоловіків віком 30-50 років Программа ИВБДВ в борьбе с диарейными заболеваниями в условиях ПМСП
Программа ИВБДВ в борьбе с диарейными заболеваниями в условиях ПМСП Выделительная система
Выделительная система Бронхиальная астма у детей раннего возраста
Бронхиальная астма у детей раннего возраста Диспансеризация и профилактические медицинские осмотры в целях раннего выявления ХНИЗ и факторов риска
Диспансеризация и профилактические медицинские осмотры в целях раннего выявления ХНИЗ и факторов риска Тактична медицина. Головні принципи надання допомоги під час бойових дій
Тактична медицина. Головні принципи надання допомоги під час бойових дій Психотропты заттар
Психотропты заттар Тері физиологиясы
Тері физиологиясы Этиология нарушений речи
Этиология нарушений речи Опиоидные (наркотические) анальгетики
Опиоидные (наркотические) анальгетики Альтернатива заместительной гормонотерапии 2014
Альтернатива заместительной гормонотерапии 2014 Дәрігердің басшылық қасиеттері және олардың кәсіптік қызметтегі алатын орны
Дәрігердің басшылық қасиеттері және олардың кәсіптік қызметтегі алатын орны Принципы лечения основных офтальмологических синдромов
Принципы лечения основных офтальмологических синдромов Электрокардиограмма
Электрокардиограмма Протокол забора биоматериала
Протокол забора биоматериала Электрдің, электромагниттік толқындардың, иондаушы және ультрадыбыстық сәулелердің адам денсаулығына әсері
Электрдің, электромагниттік толқындардың, иондаушы және ультрадыбыстық сәулелердің адам денсаулығына әсері Вирусные гепатиты при беременности
Вирусные гепатиты при беременности Ювенильный ревматоидный артрит
Ювенильный ревматоидный артрит Рахитпен (мешел) ауыратын балаларда Д витаминінің әсері
Рахитпен (мешел) ауыратын балаларда Д витаминінің әсері Производственная вибрация. Вибрационная болезнь
Производственная вибрация. Вибрационная болезнь Сочетание рентгеновских синдромов патологии легких. Рентгенопульмонология
Сочетание рентгеновских синдромов патологии легких. Рентгенопульмонология История зарождения и становления анатомии в зарубежных странах
История зарождения и становления анатомии в зарубежных странах Организационно-методическая и лечебная помощь при тяжелой пневмонии, вызванной вирусом гриппа А (H1N1)
Организационно-методическая и лечебная помощь при тяжелой пневмонии, вызванной вирусом гриппа А (H1N1) Адамдағы циркадианды ырғақтар. Адамдағы инфрадианды және ультрадианды ырғақтар. Биологиялық ырғақтардың пейсмекерлері
Адамдағы циркадианды ырғақтар. Адамдағы инфрадианды және ультрадианды ырғақтар. Биологиялық ырғақтардың пейсмекерлері Тесты функциональной диагностики
Тесты функциональной диагностики Күйзеліс туралы Г.Сельенің ілімі. Эмоциялық стресс
Күйзеліс туралы Г.Сельенің ілімі. Эмоциялық стресс Методы обеспечения (восстановления) проходимости дыхательных путей
Методы обеспечения (восстановления) проходимости дыхательных путей Острые нарушения мозгового кровообращения. Ишемический инсульт. Транзиторная ишемическая атака
Острые нарушения мозгового кровообращения. Ишемический инсульт. Транзиторная ишемическая атака