- Характеристика и классификация хромосомных болезней

Содержание
- 2. аминокислотного обмена: - Фенилкетонурия (ФКУ) - Альбинизм углеводного обмена - Галактоземия липидного обмена - Тея-Сакса -
- 3. 3. Характеристика и классификация хромосомных болезней. 4. Болезни, обусловленные изменением структуры хромосом: синдром «кошачьего крика» хронический
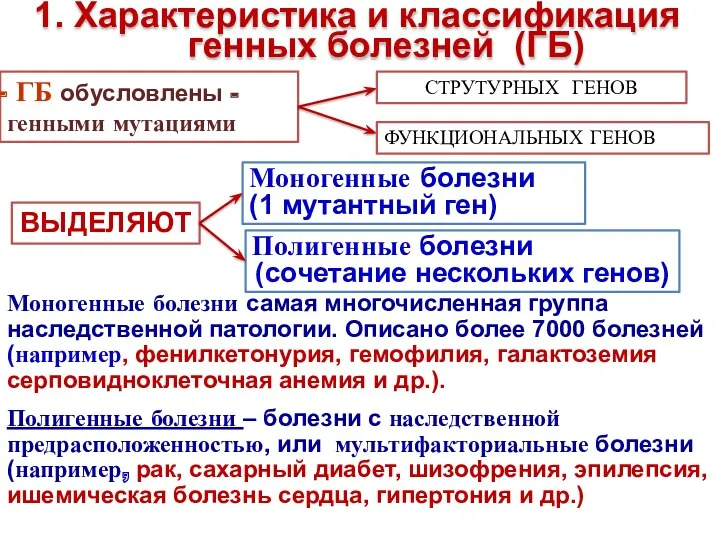
- 4. 1. Характеристика и классификация генных болезней (ГБ) ГБ обусловлены - генными мутациями СТРУТУРНЫХ ГЕНОВ ФУНКЦИОНАЛЬНЫХ ГЕНОВ
- 5. Генные болезни – это болезни, вызываемые генными мутациями, т.е. молекулярными изменениями на уровне ДНК. Генные болезни
- 6. Эти нарушения могут осуществляться на различных уровнях: претранскрипционном (уменьшение или увеличение числа копий генов), транскрипционном (генетические
- 7. Механизм возникновения генных болезней ГЕН в организме реализуется по такой системе: БИОХИМИЧЕСКАЯ РЕАКЦИЯ ГЕН ФЕРМЕНТ Повреждение
- 8. 1. Нарушения аминокислотного обмена 1. Фенилкетонурия (ФКУ) встречается с частотой 1:6000-1000, наследуется по аутосомно-рецессивному типу; больные
- 9. При нарушении активности фермента В НОРМЕ биохимическая реакция протекает по схеме: Фенилаланин Тирозин Меланин Фенилаланин гидроксилаза
- 10. Тип наследования - АР
- 12. В НОРМЕ биохимическая реакция протекает по схеме: Фенилаланин Тирозин Меланин Фенилаланин гидроксилаза Тирозиназа ФКУ АЛЬБИНИЗМ фермент
- 13. Дети с фенилкетонурией рождаются здоровыми, но в первые месяцы в связи с поступлением фенилаланина с молоком
- 14. Фенотипические проявления «Поза портного» Характерен «мышиный запах», Умственная отсталость, микроцефалия, светлые волосы и глаза
- 15. осуществляется биохимическими методами: в моче ФПВК, в крови – высокое содержание фенилаланина. Возможна пренатальная диагностика (амниоцентез
- 16. При этом заболевании в коже не образуется пигмент меланин. Первичным биохимическим дефектом является недостаточность фермента тирозиназы,
- 17. Альбинизм наследуется по аутосомно-рецессивному типу
- 18. Основным клиническим проявлением альбинизма в любом возрасте являются отсутствие меланина в клетках кожи (молочно-белый ее цвет),
- 19. 2. Нарушения углеводного обмена Галактоземия Заболевание связано с отсутствием или резким снижением активности фермента галактозо-1-фосфатуридилтрансферазы (Г-1-ФУТ),
- 20. больные (аа) 25% Тип наследования АР
- 21. Лактоза — главный углевод молока — представляет собой дисахарид, содержащий галактозу и глюкозу. В пищеварительных маршрутах
- 22. На следующем этапе галактозо-1-фосфат превращается в глюкозо-1-фосфат под действием фермента ГАЛТ, ген которого расположен на 9-й
- 23. При недостаточности галактокиназы галактоза накапливается в крови и тканях. В хрусталике глаза она под действием альдозоредуктазы
- 24. У новорожденных: желтуха, судороги, рвота, диарея, умственная отсталость. Без лечения больные погибают, у выживших – умственная
- 25. Галактоземия Повреждение головного мозга Катаракта Желтуха Цирроз печени Повреждение почек
- 26. 3. Нарушения липидного обмена БолезньТея-Сакса Частота встречаемости 1:250 000 среди евреев-ашкенази 1:4 000 смерть наступает в
- 27. Заболевание вызвано мутацией в гене HEXA, который кодирует α-субъединицу фермента гексозоаминидазы A и находится на длинном
- 28. Наследуется по аутосомно-рецессивному типу с частотой 1:250000, а среди евреев-ашкинази – 1:3600 новорожденных. Новорождённые с данным
- 29. атрофия зрительных нервов, слепота, слабоумие, на глазном дне симптом вишневой косточки, Фенотипические проявления: Заболевание развивается медленно:
- 30. Лабораторная диагностика: Биохимические реакции
- 31. Наследственная доминантная болезнь соединительной ткани. Синдром клинически идентифицировал В. Марфан в 1886 г. Причиной синдрома Марфана
- 32. Наиболее специфичными для синдрома Марфана являются нарушения скелета, вывих хрусталика, сердечно-сосудистые изменения, эктазия твердой мозговой оболочки.
- 33. Тест Синдром Марфана
- 34. Синдром Марфана Готическое небо Вывиххрусталика
- 35. Недостаток белка церуллоплазмина, который содержит медь Тип наследования: АР. Частота встречаемости : 1:50 000. Гепатоцеребральная дистрофия
- 36. Фенотипические проявления болезни Вильсона-Коновалова Поражение почек (синдром Фанкони) Гемолиз Остеопения Накопления меди Судороги Нейропсихические нарушения Кольцо
- 37. Биохимические реакции - определение уровня церулоплазмина (типично снижение менее 1 мкмоль/л) - определение уровня меди в
- 38. «ХРОМОСОМНЫЕ БОЛЕЗНИ»
- 39. Словарь Гипертелоризм (др.-греч. ὑπερ — чрезмерно, τῆλε — далеко, ὁρίζω — разделять) — ненормальное (увеличенное) расстояние
- 40. Эпика́нтус, «монгольская складка» — особая складка у внутреннего угла глаза, в большей или меньшей степени прикрывающая
- 41. Микроцефалия (от греч. μικρός — маленький и κεφαλή — голова) — значительное уменьшение размеров черепа и,
- 42. Спленомегалия – увеличение размеров селезенки Гепатомегалия – увеличение размеров печени Гепатоспленомегалия – увеличение размеров печени и
- 43. Мышечная гипотония (мышечный гипотонус) — состояние пониженного мышечного тонуса (степени напряжения мышцы или её сопротивления движению),
- 44. ХАРАКТЕРИСТИКА И КЛАССИФИКАЦИЯ ХРОМОСОМНЫХ БОЛЕЗНЕЙ. Хромосомные болезни - это большая группа наследственных болезней, клинически характеризующихся МНОЖЕСТВЕННЫМИ
- 45. В основу классификации хромосомных болезней положены тип хромосомной или геномной мутации и индивидуальность вовлекаемой в изменения
- 46. обусловлен частичной моносомией короткого плеча 5-й хромосомы СИНДРОМ «КОШАЧЬЕГО КРИКА» (-5р-) (46, 5р15)
- 47. Частота встречаемости – 1: 45000. За развитие синдрома ответственен небольшой участок в коротком плече 5-й хромосомы
- 48. Для данного синдрома характерен специфический плач, напоминающий кошачье мяуканье, за что так и назвали эту патологию.
- 49. Синдром кошачьего крика
- 50. Продолжительность жизни значительно снижена, большинство больных погибают в первые годы, около 10% доживают до 10-летнего возраста.
- 51. ХРОНИЧЕСКИЙ МИЕЛОЛЕЙКОЗ обусловлен транслокацией между 9-й и 21-22-й хромосомами вызывают (опухолевидное разрастание белого ростка крови). Такие
- 52. Хронический миелолейкоз
- 53. 3. БОЛЕЗНИ, ОБУСЛОВЛЕННЫЕ ИЗМЕНЕНИЕМ ЧИСЛА ХРОМОСОМ. В основе этой категории хромосомных болезней лежат мутации, связанные с
- 54. Это летальная мутация - дети умирают до рождения или в первые часы после рождения. Крайне редко
- 55. АНЕУПЛОИДИИ Составляют основную массу хромосомных болезней составляют Большинство хромосомных аномалий по крупным хромосомам не совместимы с
- 56. У человека наиболее часто встречаются трисомии по 13, 18 и 21 паре аутосом. В связи с
- 57. синдром трисомии – 13 (47,13+ и 46,13/13) встречается с частотой 1:6000. Имеются два цитогенетических варианта синдрома
- 58. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500г). Соотношение полов близко 1:1. Фенотип
- 59. Синдром Патау
- 60. Расщелина губы и неба
- 61. Полидактилия
- 62. Микроцефалия
- 64. синдром трисомии – 18 (47,18+). Характерные черты синдрома Эдвардса связаны с трисомией сегмента хромосомы 18q 11
- 66. СИНДРОМ ЭДВАРДСА
- 67. синдром трисомии 21 (47,21+; 46 21/21) Самая частая форма хромосомной патологии у человека – 1: 550-650.
- 68. Масса новорожденных с СД в среднем 3100. Соотношение полов близко 1:4. Для больных характерны низкий рост,
- 69. СИНДРОМ ДАУНА
- 70. СИНДРОМ ДАУНА
- 71. СИНДРОМ ДАУНА Поперечная борозда, Угол ATD >100 градусов Эпикант большой уплощенный язык с глубокими бороздами сандалевидный
- 72. СИНДРОМ ДАУНА
- 73. СИНДРОМ ТРИСОМИИ (9р). Синдром трисомии по короткому плечу хромосомы 9 (синдром Реторе) – часто встречающаяся форма
- 74. Моносомии по какой-либо аутосоме с жизнью не совместимы. Совместимыми с жизнью являются изменения количества половых хромосом,
- 75. СИНДРОМ ШЕРШЕВСКОГО-ТЕРНЕРА (45, ХО) единственная форма моносомии у живорожденных, однако 90% гибнут внутриутробно, а из рожденных,
- 76. СИНДРОМ ШЕРШЕВСКОГО-ТЕРНЕРА 45,ХО
- 77. СИНДРОМ ШЕРШЕВСКОГО-ТЕРНЕРА
- 79. Скачать презентацию

аминокислотного обмена:
- Фенилкетонурия (ФКУ)
- Альбинизм
углеводного обмена
- Галактоземия
липидного обмена
- Тея-Сакса
- Нимана-Пика
минерального обмена
-
аминокислотного обмена:
- Фенилкетонурия (ФКУ)
- Альбинизм
углеводного обмена
- Галактоземия
липидного обмена
- Тея-Сакса
- Нимана-Пика
минерального обмена
-

3. Характеристика и классификация хромосомных болезней.
4. Болезни, обусловленные изменением структуры хромосом:
синдром
3. Характеристика и классификация хромосомных болезней.
4. Болезни, обусловленные изменением структуры хромосом:
синдром
1. Характеристика и классификация
генных болезней (ГБ)
ГБ обусловлены -
генными
1. Характеристика и классификация
генных болезней (ГБ)
ГБ обусловлены -
генными

Генные болезни – это болезни, вызываемые генными мутациями, т.е. молекулярными изменениями
Генные болезни – это болезни, вызываемые генными мутациями, т.е. молекулярными изменениями

Эти нарушения могут осуществляться на различных уровнях:
претранскрипционном (уменьшение или увеличение
Эти нарушения могут осуществляться на различных уровнях:
претранскрипционном (уменьшение или увеличение
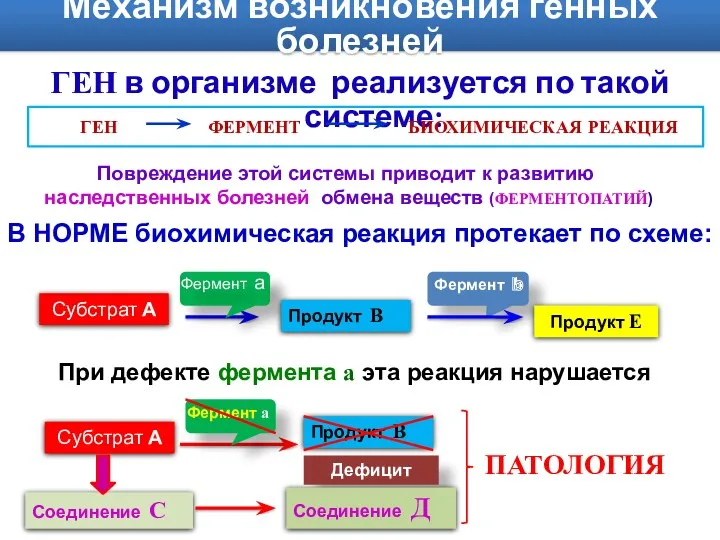
Механизм возникновения генных болезней
ГЕН в организме реализуется по такой системе:
БИОХИМИЧЕСКАЯ РЕАКЦИЯ
Механизм возникновения генных болезней
ГЕН в организме реализуется по такой системе:
БИОХИМИЧЕСКАЯ РЕАКЦИЯ

1. Нарушения аминокислотного обмена
1. Фенилкетонурия (ФКУ)
встречается с частотой 1:6000-1000, наследуется по
1. Нарушения аминокислотного обмена
1. Фенилкетонурия (ФКУ)
встречается с частотой 1:6000-1000, наследуется по
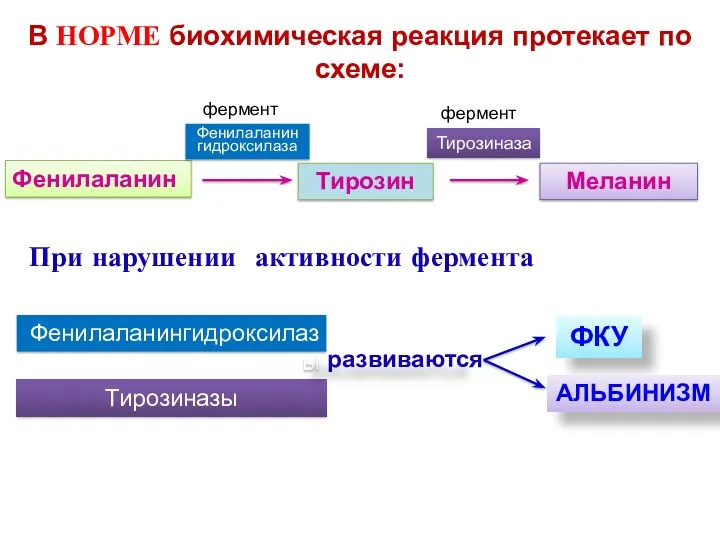
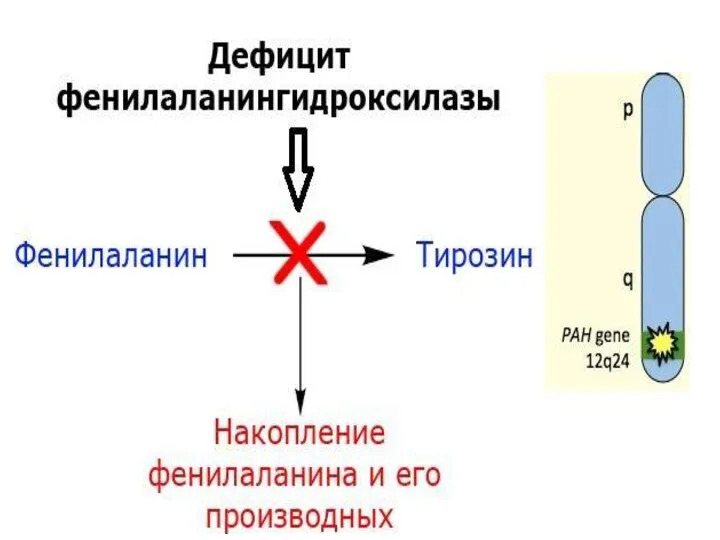
При нарушении активности фермента
В НОРМЕ биохимическая реакция протекает по схеме:
При нарушении активности фермента
В НОРМЕ биохимическая реакция протекает по схеме:
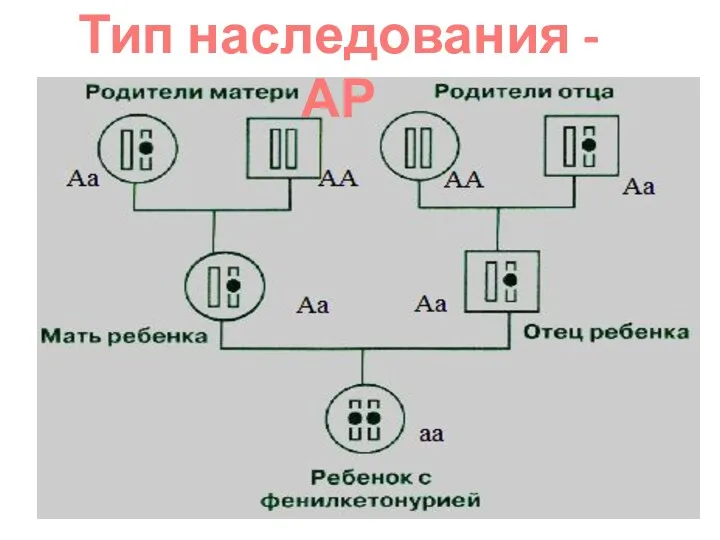
Тип наследования - АР
Тип наследования - АР
В НОРМЕ биохимическая реакция протекает по схеме:
Фенилаланин
Тирозин
Меланин
Фенилаланин
гидроксилаза
Тирозиназа
ФКУ
АЛЬБИНИЗМ
фермент
фермент
В Донецке 1 :
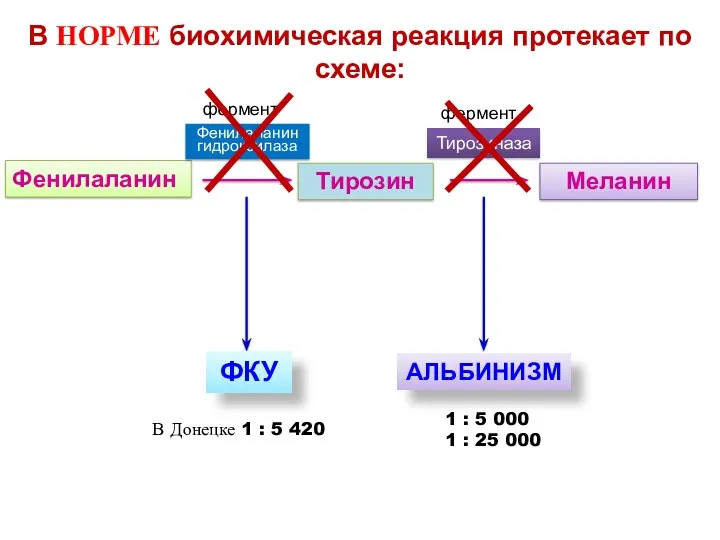
В НОРМЕ биохимическая реакция протекает по схеме:
Фенилаланин
Тирозин
Меланин
Фенилаланин
гидроксилаза
Тирозиназа
ФКУ
АЛЬБИНИЗМ
фермент
фермент
В Донецке 1 :

Дети с фенилкетонурией рождаются здоровыми, но в первые месяцы в связи
Дети с фенилкетонурией рождаются здоровыми, но в первые месяцы в связи

Фенотипические проявления
«Поза портного»
Характерен «мышиный запах»,
Умственная отсталость, микроцефалия,
светлые волосы и
Фенотипические проявления
«Поза портного»
Характерен «мышиный запах»,
Умственная отсталость, микроцефалия,
светлые волосы и
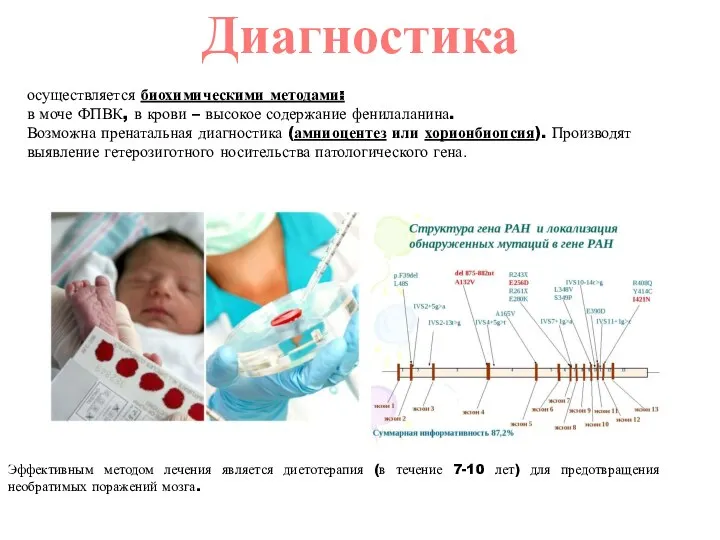
осуществляется биохимическими методами:
в моче ФПВК, в крови – высокое содержание
осуществляется биохимическими методами:
в моче ФПВК, в крови – высокое содержание
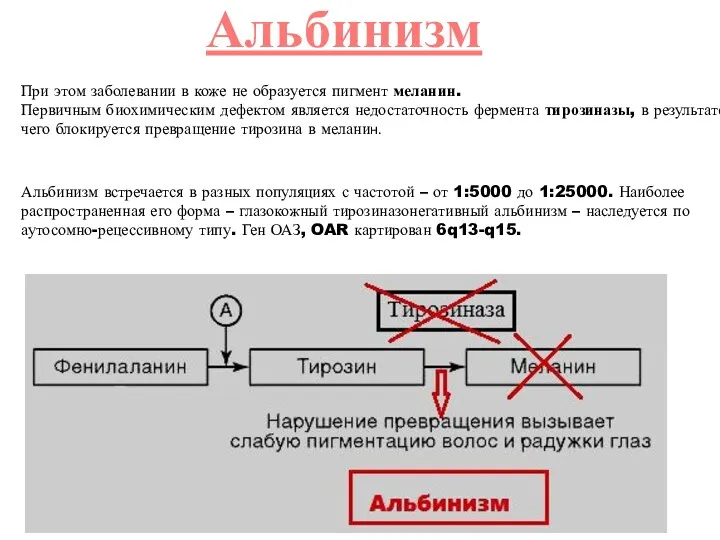
При этом заболевании в коже не образуется пигмент меланин.
Первичным биохимическим
При этом заболевании в коже не образуется пигмент меланин.
Первичным биохимическим
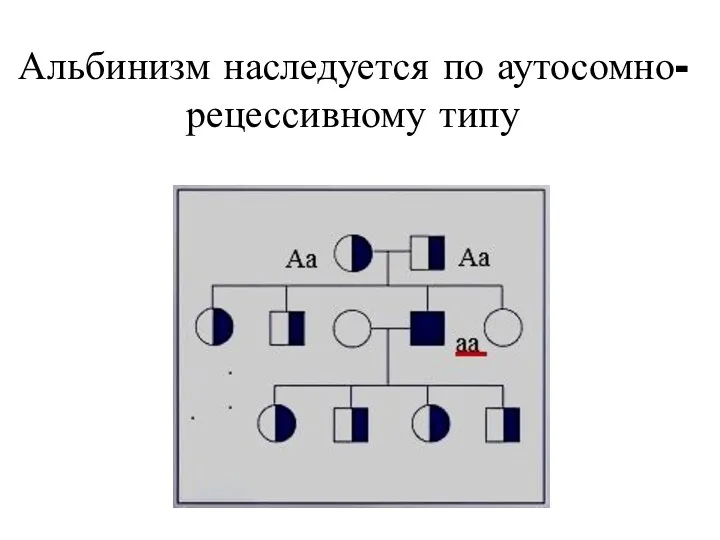
Альбинизм наследуется по аутосомно-рецессивному типу
Альбинизм наследуется по аутосомно-рецессивному типу

Основным клиническим проявлением альбинизма в любом возрасте являются отсутствие меланина в
Основным клиническим проявлением альбинизма в любом возрасте являются отсутствие меланина в

2. Нарушения углеводного обмена
Галактоземия
Заболевание связано с отсутствием или резким снижением активности
2. Нарушения углеводного обмена
Галактоземия
Заболевание связано с отсутствием или резким снижением активности

больные (аа) 25%
Тип наследования АР
больные (аа) 25%
Тип наследования АР
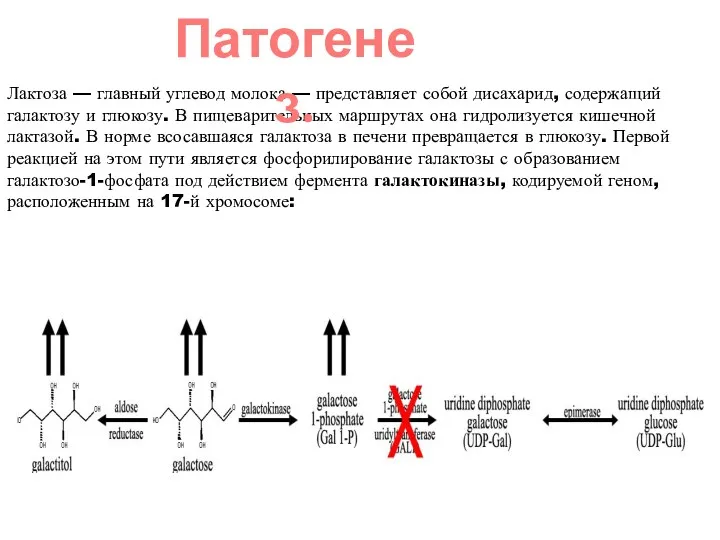
Лактоза — главный углевод молока — представляет собой дисахарид, содержащий галактозу
Лактоза — главный углевод молока — представляет собой дисахарид, содержащий галактозу

На следующем этапе галактозо-1-фосфат превращается в глюкозо-1-фосфат под действием фермента ГАЛТ,
На следующем этапе галактозо-1-фосфат превращается в глюкозо-1-фосфат под действием фермента ГАЛТ,

При недостаточности галактокиназы галактоза накапливается в крови и тканях. В хрусталике
При недостаточности галактокиназы галактоза накапливается в крови и тканях. В хрусталике
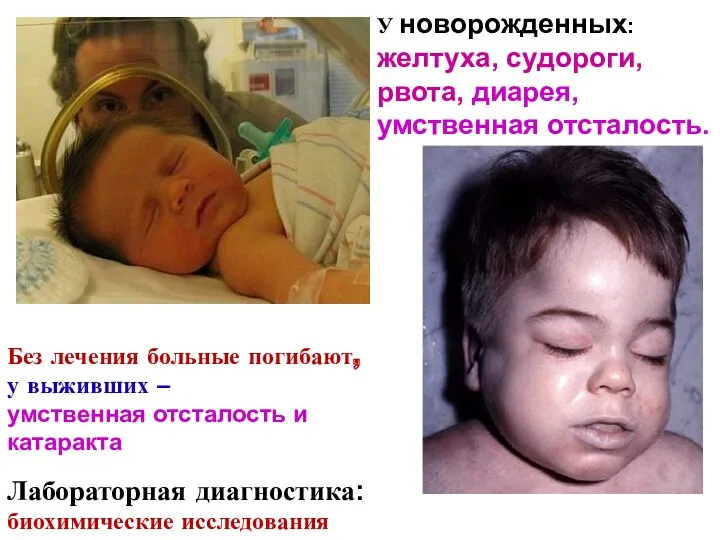
У новорожденных:
желтуха, судороги,
рвота, диарея, умственная отсталость.
Без лечения больные погибают,
у выживших
У новорожденных:
желтуха, судороги,
рвота, диарея, умственная отсталость.
Без лечения больные погибают,
у выживших
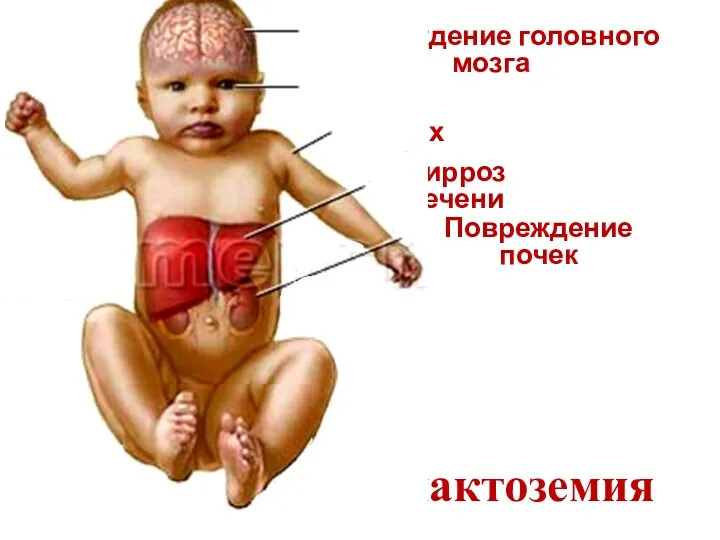
Галактоземия
Повреждение головного мозга
Катаракта
Желтуха
Цирроз печени
Повреждение почек
Галактоземия
Повреждение головного мозга
Катаракта
Желтуха
Цирроз печени
Повреждение почек

3. Нарушения липидного обмена
БолезньТея-Сакса
Частота встречаемости
1:250 000
среди евреев-ашкенази
1:4 000
смерть наступает
3. Нарушения липидного обмена
БолезньТея-Сакса
Частота встречаемости
1:250 000
среди евреев-ашкенази
1:4 000
смерть наступает
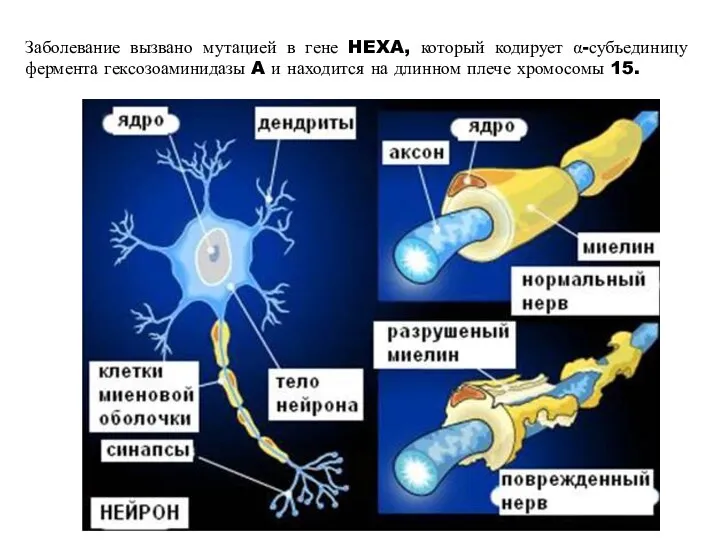
Заболевание вызвано мутацией в гене HEXA, который кодирует α-субъединицу фермента гексозоаминидазы
Заболевание вызвано мутацией в гене HEXA, который кодирует α-субъединицу фермента гексозоаминидазы

Наследуется по аутосомно-рецессивному типу с частотой 1:250000,
а среди евреев-ашкинази –
Наследуется по аутосомно-рецессивному типу с частотой 1:250000,
а среди евреев-ашкинази –
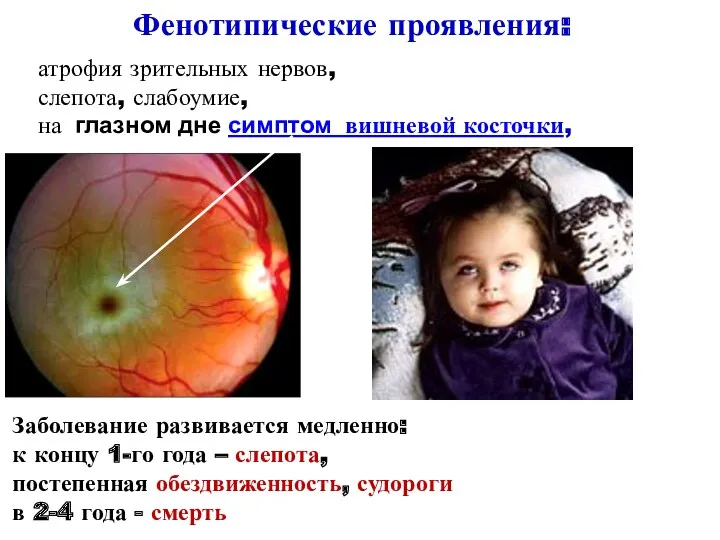
атрофия зрительных нервов,
слепота, слабоумие,
на глазном дне симптом вишневой косточки,
атрофия зрительных нервов,
слепота, слабоумие,
на глазном дне симптом вишневой косточки,

Лабораторная диагностика:
Биохимические реакции
Лабораторная диагностика:
Биохимические реакции

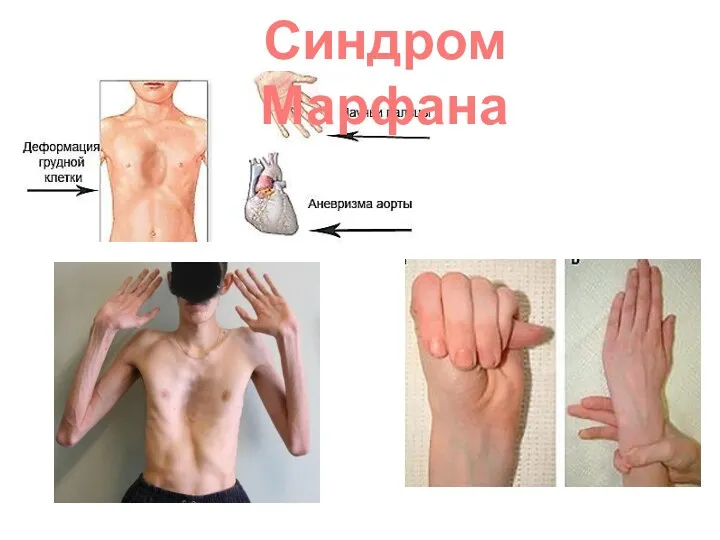
Наследственная доминантная болезнь соединительной ткани. Синдром клинически идентифицировал В. Марфан в
Наследственная доминантная болезнь соединительной ткани. Синдром клинически идентифицировал В. Марфан в

Наиболее специфичными для синдрома Марфана являются нарушения скелета, вывих хрусталика, сердечно-сосудистые
Наиболее специфичными для синдрома Марфана являются нарушения скелета, вывих хрусталика, сердечно-сосудистые
Тест
Синдром Марфана
Тест
Синдром Марфана
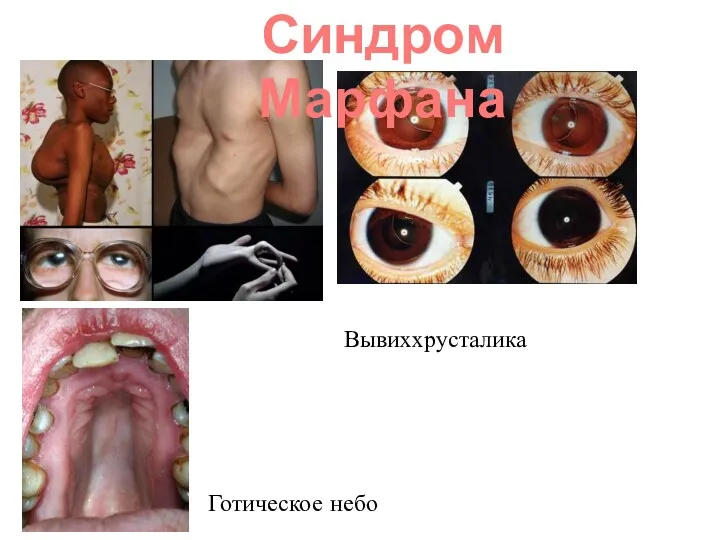
Синдром Марфана
Готическое небо
Вывиххрусталика
Синдром Марфана
Готическое небо
Вывиххрусталика

Недостаток белка церуллоплазмина,
который содержит медь
Тип наследования: АР.
Частота встречаемости :
1:50 000.
Гепатоцеребральная
Недостаток белка церуллоплазмина,
который содержит медь
Тип наследования: АР.
Частота встречаемости :
1:50 000.
Гепатоцеребральная
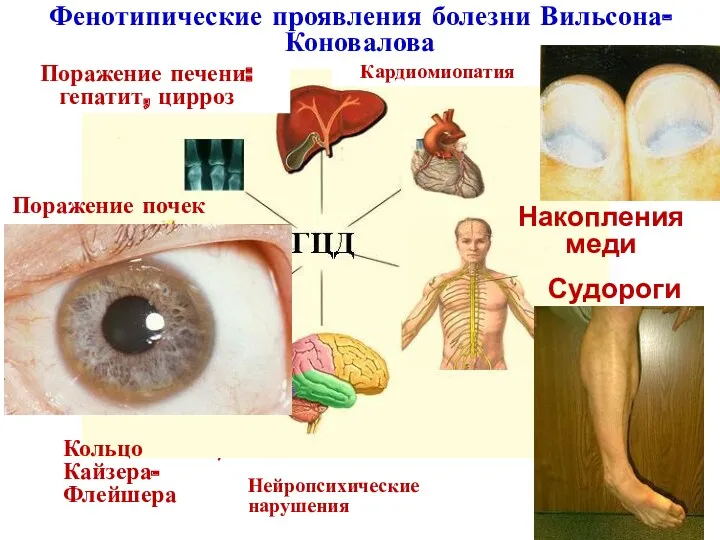
Фенотипические проявления болезни Вильсона-Коновалова
Поражение почек
(синдром Фанкони)
Гемолиз
Остеопения
Накопления
меди
Судороги
Нейропсихические
нарушения
Кольцо
Кайзера-
Флейшера
Кардиомиопатия
Поражение печени:
гепатит,
Фенотипические проявления болезни Вильсона-Коновалова
Поражение почек
(синдром Фанкони)
Гемолиз
Остеопения
Накопления
меди
Судороги
Нейропсихические
нарушения
Кольцо
Кайзера-
Флейшера
Кардиомиопатия
Поражение печени:
гепатит,
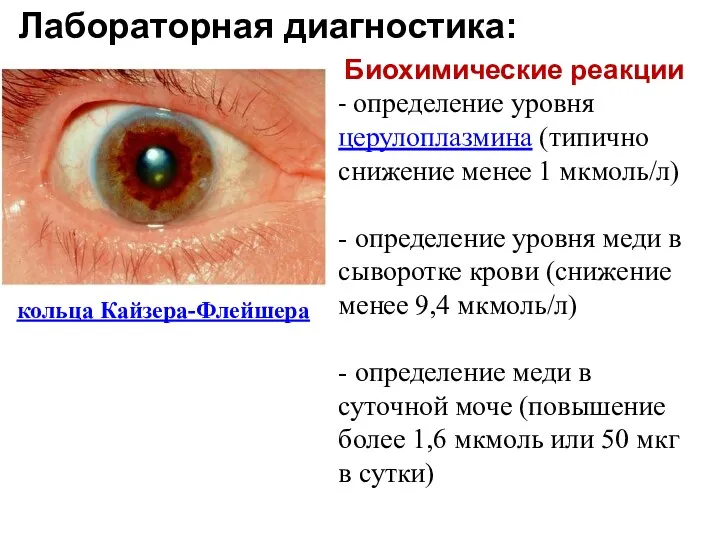
Биохимические реакции
- определение уровня церулоплазмина (типично снижение менее 1 мкмоль/л)
- определение
Биохимические реакции
- определение уровня церулоплазмина (типично снижение менее 1 мкмоль/л)
- определение
«ХРОМОСОМНЫЕ БОЛЕЗНИ»
«ХРОМОСОМНЫЕ БОЛЕЗНИ»

Словарь
Гипертелоризм (др.-греч. ὑπερ — чрезмерно, τῆλε — далеко, ὁρίζω — разделять)
Словарь
Гипертелоризм (др.-греч. ὑπερ — чрезмерно, τῆλε — далеко, ὁρίζω — разделять)
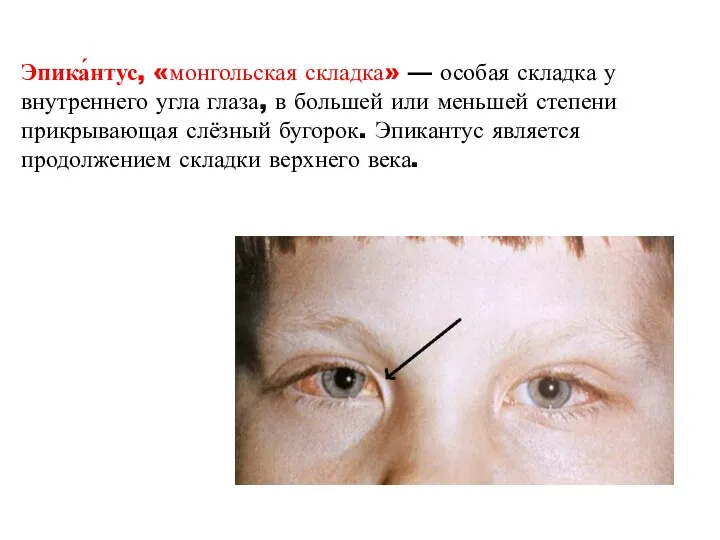
Эпика́нтус, «монгольская складка» — особая складка у внутреннего угла глаза, в
Эпика́нтус, «монгольская складка» — особая складка у внутреннего угла глаза, в
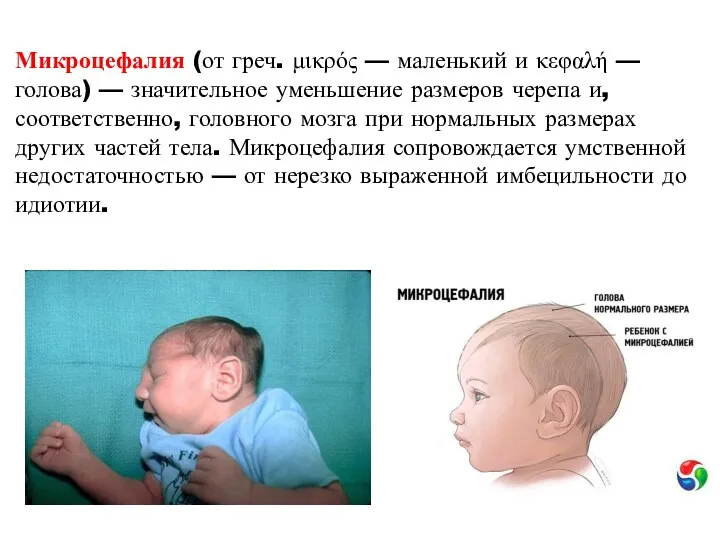
Микроцефалия (от греч. μικρός — маленький и κεφαλή — голова) —
Микроцефалия (от греч. μικρός — маленький и κεφαλή — голова) —
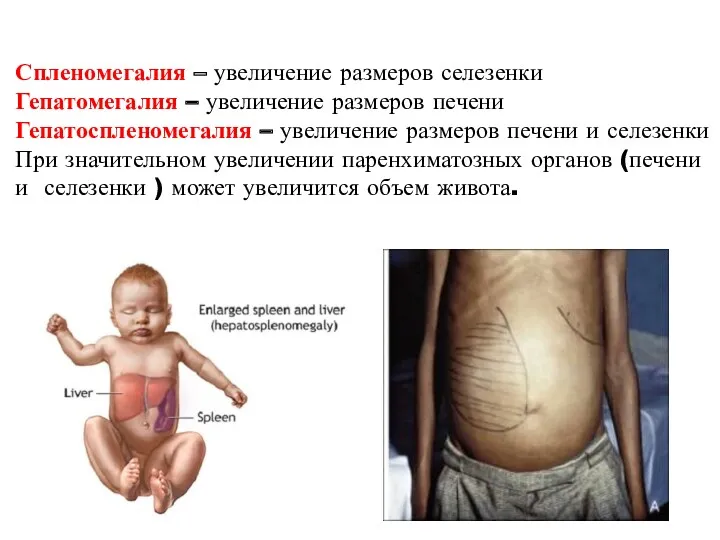
Спленомегалия – увеличение размеров селезенки
Гепатомегалия – увеличение размеров печени
Гепатоспленомегалия – увеличение
Спленомегалия – увеличение размеров селезенки
Гепатомегалия – увеличение размеров печени
Гепатоспленомегалия – увеличение
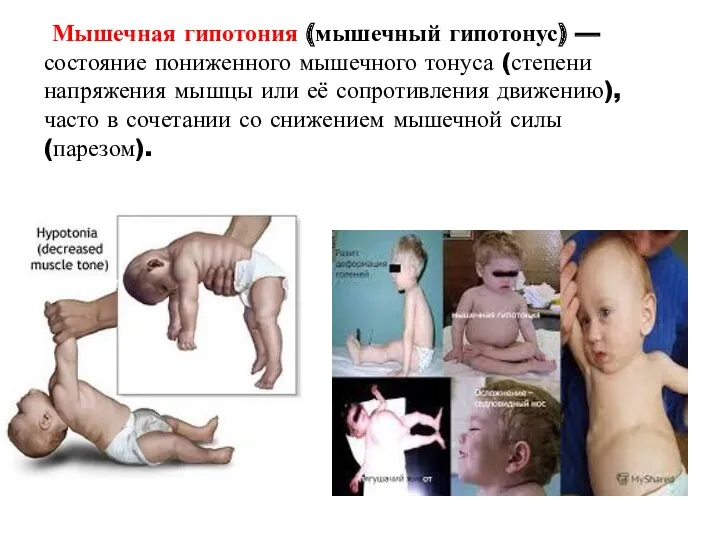
Мышечная гипотония (мышечный гипотонус) — состояние пониженного мышечного тонуса (степени
Мышечная гипотония (мышечный гипотонус) — состояние пониженного мышечного тонуса (степени

ХАРАКТЕРИСТИКА И КЛАССИФИКАЦИЯ ХРОМОСОМНЫХ БОЛЕЗНЕЙ.
Хромосомные болезни - это большая группа
ХАРАКТЕРИСТИКА И КЛАССИФИКАЦИЯ ХРОМОСОМНЫХ БОЛЕЗНЕЙ.
Хромосомные болезни - это большая группа

В основу классификации хромосомных болезней
положены тип хромосомной или геномной
В основу классификации хромосомных болезней
положены тип хромосомной или геномной
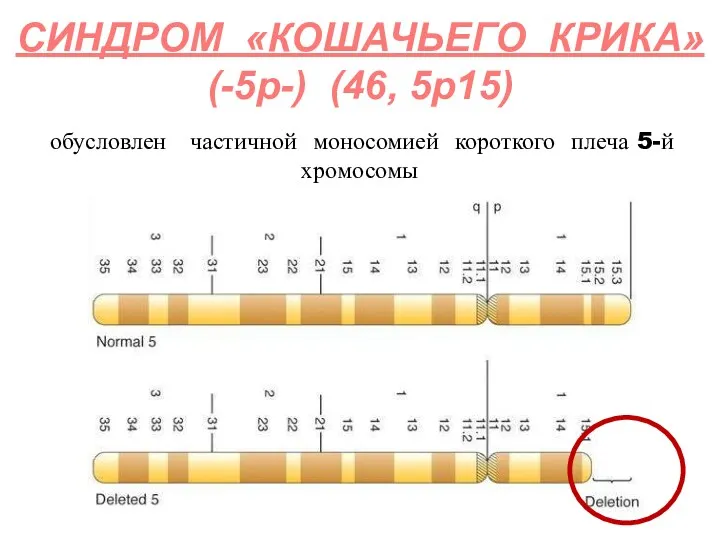
обусловлен частичной моносомией короткого плеча 5-й хромосомы
СИНДРОМ «КОШАЧЬЕГО КРИКА»
(-5р-)
обусловлен частичной моносомией короткого плеча 5-й хромосомы
СИНДРОМ «КОШАЧЬЕГО КРИКА»
(-5р-)

Частота встречаемости – 1: 45000. За развитие синдрома ответственен небольшой участок
Частота встречаемости – 1: 45000. За развитие синдрома ответственен небольшой участок

Для данного синдрома характерен специфический плач, напоминающий кошачье мяуканье, за что
Для данного синдрома характерен специфический плач, напоминающий кошачье мяуканье, за что

Синдром кошачьего крика
Синдром кошачьего крика

Продолжительность жизни значительно снижена, большинство больных погибают в первые годы, около
Продолжительность жизни значительно снижена, большинство больных погибают в первые годы, около

ХРОНИЧЕСКИЙ МИЕЛОЛЕЙКОЗ обусловлен транслокацией между 9-й и 21-22-й хромосомами вызывают (опухолевидное
ХРОНИЧЕСКИЙ МИЕЛОЛЕЙКОЗ обусловлен транслокацией между 9-й и 21-22-й хромосомами вызывают (опухолевидное
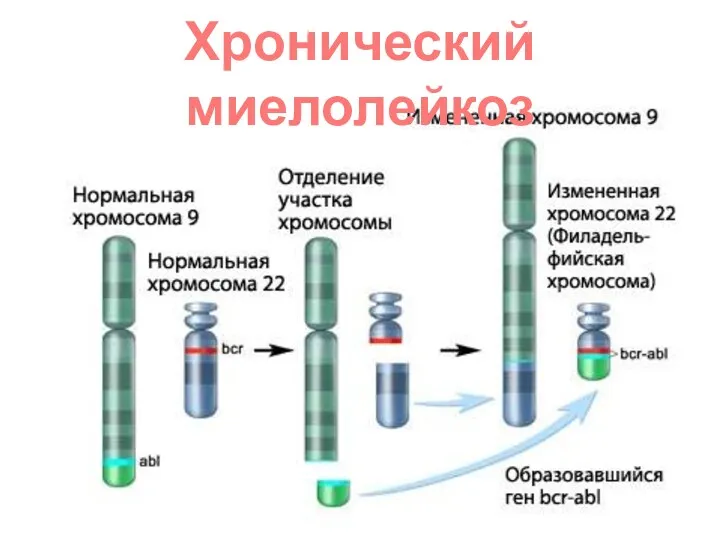
Хронический миелолейкоз
Хронический миелолейкоз

3. БОЛЕЗНИ, ОБУСЛОВЛЕННЫЕ ИЗМЕНЕНИЕМ ЧИСЛА ХРОМОСОМ.
В основе этой категории
3. БОЛЕЗНИ, ОБУСЛОВЛЕННЫЕ ИЗМЕНЕНИЕМ ЧИСЛА ХРОМОСОМ.
В основе этой категории
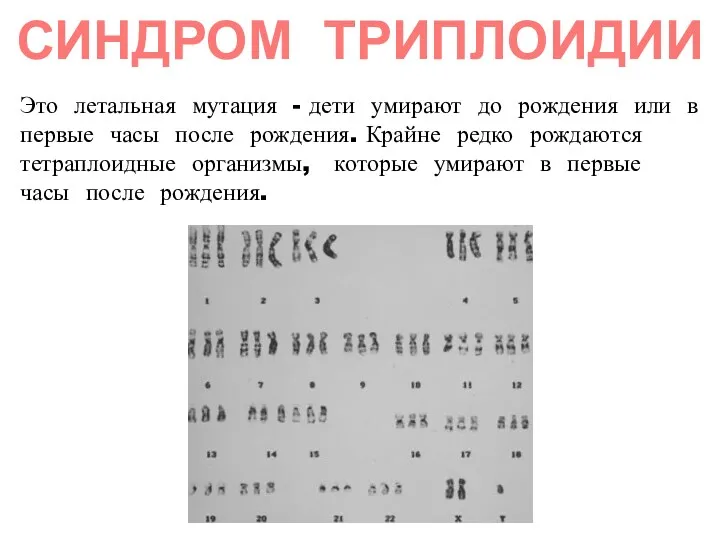
Это летальная мутация - дети умирают до рождения или в первые
Это летальная мутация - дети умирают до рождения или в первые

АНЕУПЛОИДИИ
Составляют основную массу хромосомных болезней составляют
Большинство хромосомных аномалий по крупным
АНЕУПЛОИДИИ
Составляют основную массу хромосомных болезней составляют
Большинство хромосомных аномалий по крупным

У человека наиболее часто встречаются трисомии по 13, 18 и 21
У человека наиболее часто встречаются трисомии по 13, 18 и 21
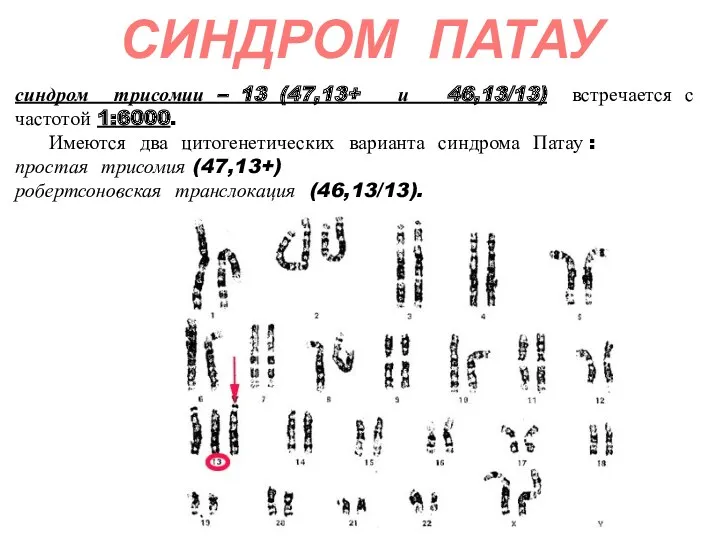
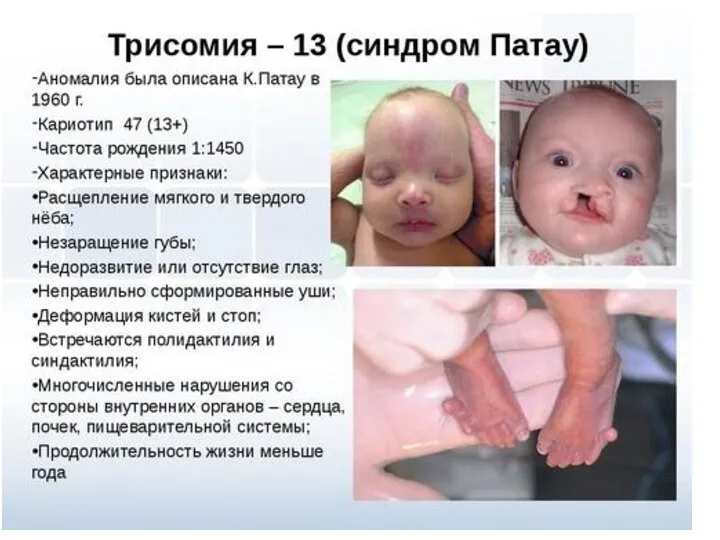
синдром трисомии – 13 (47,13+ и 46,13/13) встречается с частотой 1:6000.
синдром трисомии – 13 (47,13+ и 46,13/13) встречается с частотой 1:6000.

Дети с синдромом Патау рождаются с массой тела ниже нормы (2500г).
Дети с синдромом Патау рождаются с массой тела ниже нормы (2500г).
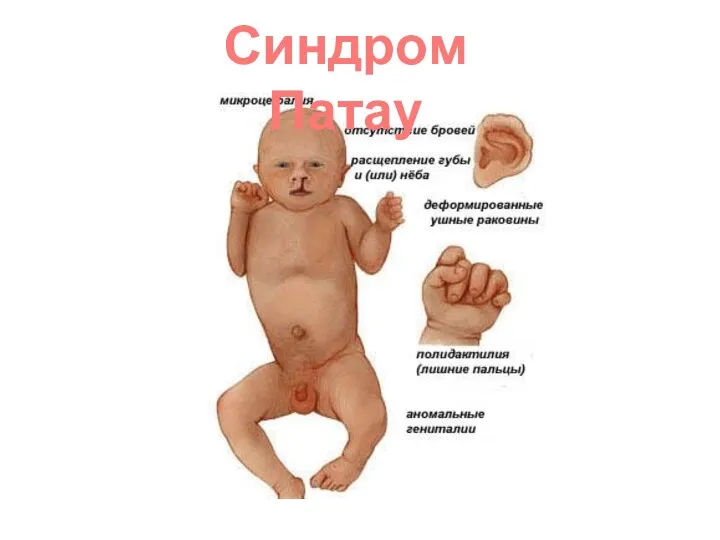
Синдром Патау
Синдром Патау
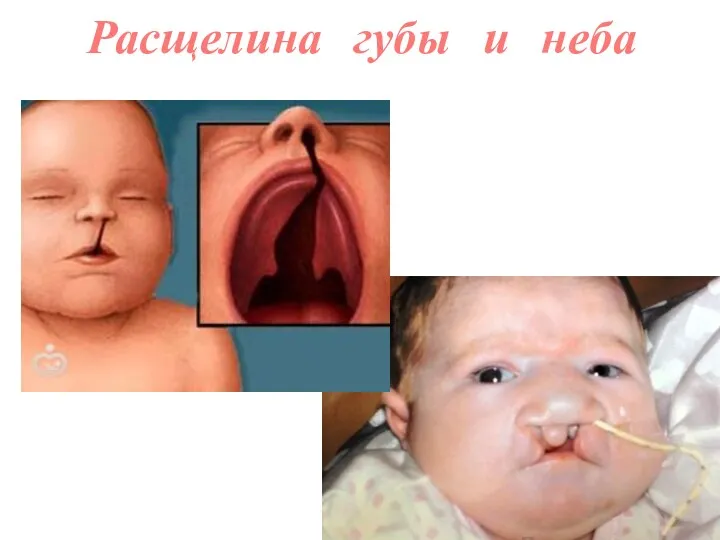
Расщелина губы и неба
Расщелина губы и неба
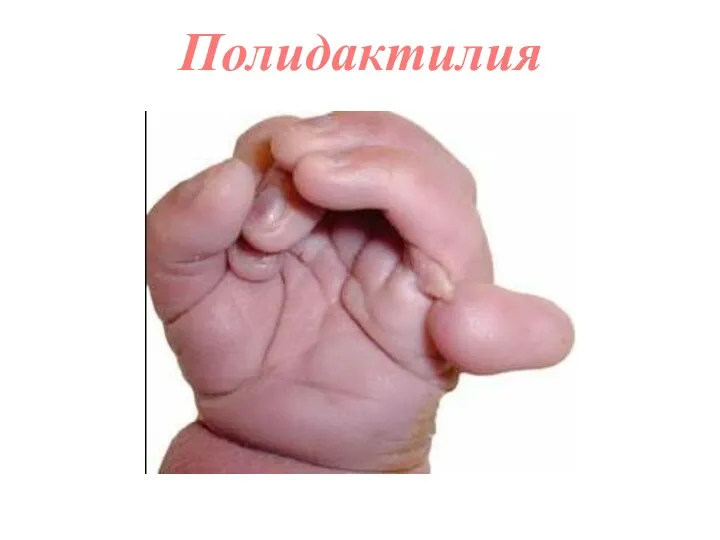
Полидактилия
Полидактилия
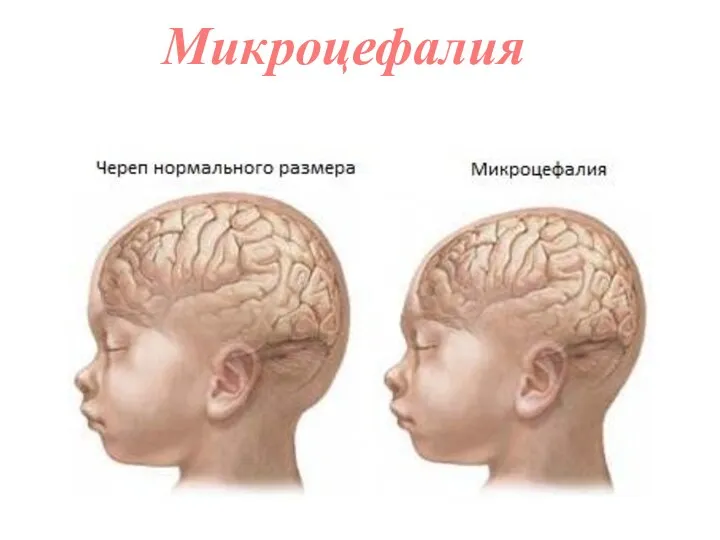
Микроцефалия
Микроцефалия

синдром трисомии – 18 (47,18+). Характерные черты синдрома Эдвардса связаны с
синдром трисомии – 18 (47,18+). Характерные черты синдрома Эдвардса связаны с
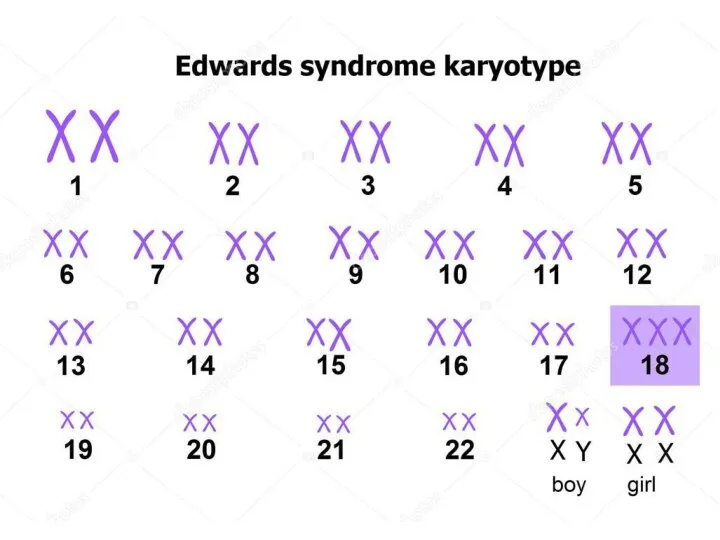
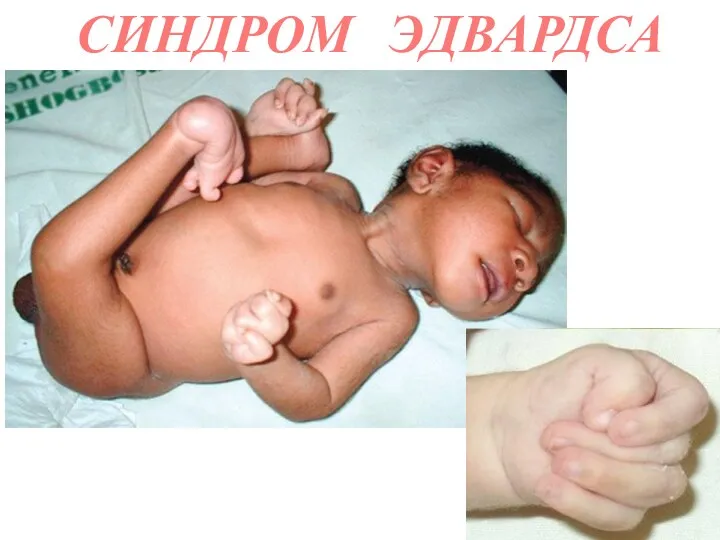
СИНДРОМ ЭДВАРДСА
СИНДРОМ ЭДВАРДСА

синдром трисомии 21 (47,21+; 46 21/21)
Самая частая форма хромосомной
синдром трисомии 21 (47,21+; 46 21/21)
Самая частая форма хромосомной

Масса новорожденных с СД в среднем 3100. Соотношение полов близко
Масса новорожденных с СД в среднем 3100. Соотношение полов близко

СИНДРОМ ДАУНА
СИНДРОМ ДАУНА

СИНДРОМ ДАУНА
СИНДРОМ ДАУНА
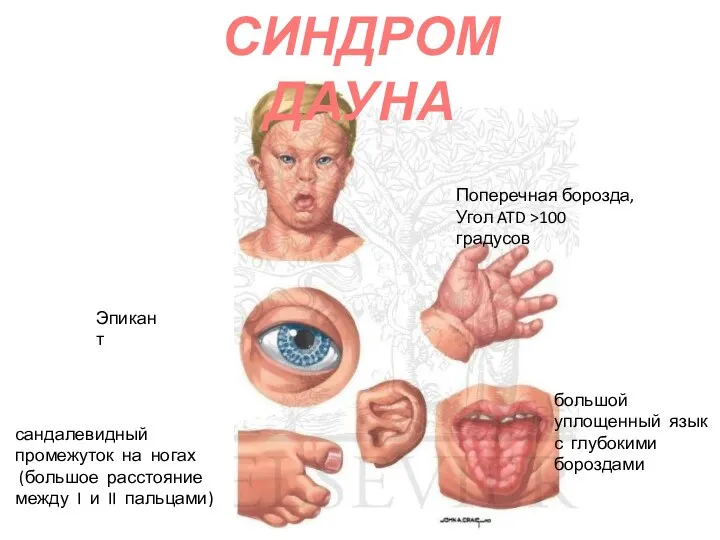
СИНДРОМ ДАУНА
Поперечная борозда,
Угол ATD >100 градусов
Эпикант
большой уплощенный язык с
СИНДРОМ ДАУНА
Поперечная борозда,
Угол ATD >100 градусов
Эпикант
большой уплощенный язык с
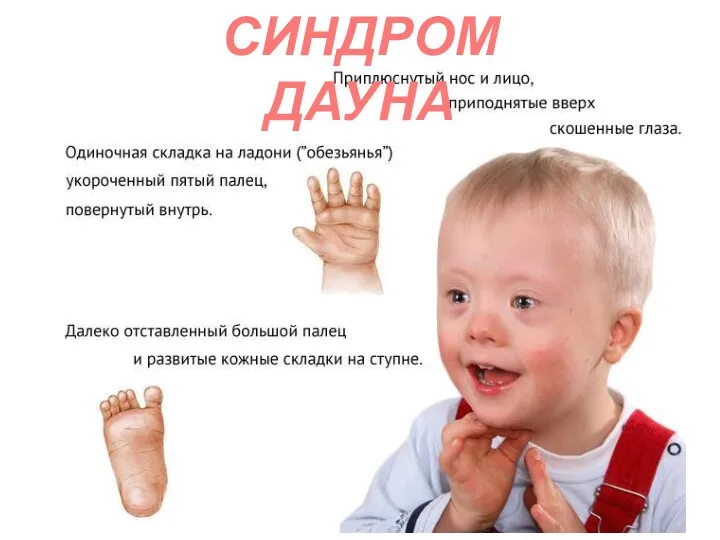
СИНДРОМ ДАУНА
СИНДРОМ ДАУНА

СИНДРОМ ТРИСОМИИ (9р).
Синдром трисомии по короткому плечу хромосомы 9
СИНДРОМ ТРИСОМИИ (9р).
Синдром трисомии по короткому плечу хромосомы 9

Моносомии по какой-либо аутосоме с жизнью не совместимы. Совместимыми с жизнью
Моносомии по какой-либо аутосоме с жизнью не совместимы. Совместимыми с жизнью

СИНДРОМ ШЕРШЕВСКОГО-ТЕРНЕРА (45, ХО) единственная форма моносомии у живорожденных, однако 90%
СИНДРОМ ШЕРШЕВСКОГО-ТЕРНЕРА (45, ХО) единственная форма моносомии у живорожденных, однако 90%
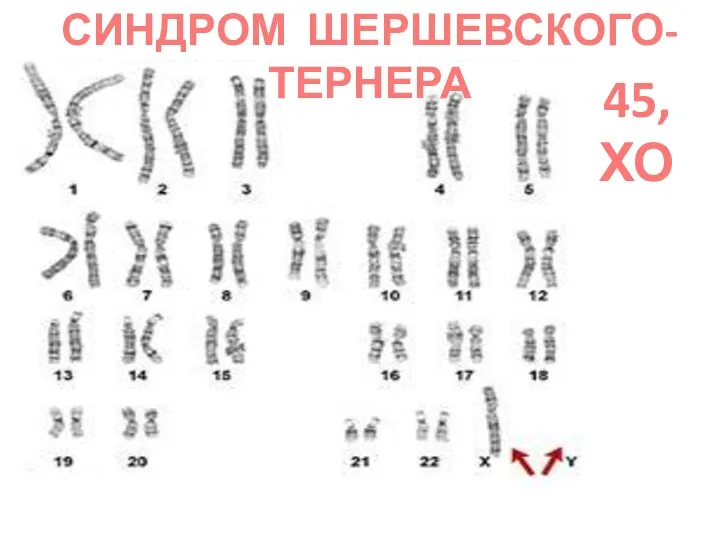
СИНДРОМ ШЕРШЕВСКОГО-ТЕРНЕРА
45,ХО
СИНДРОМ ШЕРШЕВСКОГО-ТЕРНЕРА
45,ХО
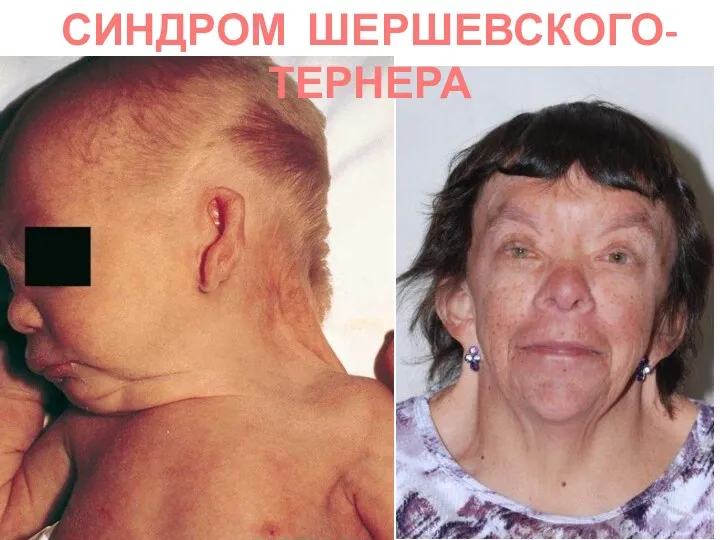
СИНДРОМ ШЕРШЕВСКОГО-ТЕРНЕРА
СИНДРОМ ШЕРШЕВСКОГО-ТЕРНЕРА
 Энтеральное и парентеральное питание в онкологии. Принципы лечение хронического болевого синдрома
Энтеральное и парентеральное питание в онкологии. Принципы лечение хронического болевого синдрома Искусственный интеллект в медицине
Искусственный интеллект в медицине Заболевания кожи. Заболевания ногтей
Заболевания кожи. Заболевания ногтей Риносинусогенді орбитальды асқынулар
Риносинусогенді орбитальды асқынулар Криотерапия. Механизмы лечебных эффектов
Криотерапия. Механизмы лечебных эффектов Клинический случай. Менингококковая инфекция. Комбинированная форма
Клинический случай. Менингококковая инфекция. Комбинированная форма Острый респираторный дистресс-синдром
Острый респираторный дистресс-синдром Случай из практики
Случай из практики Инфекционная безопасность пациентов и персонала
Инфекционная безопасность пациентов и персонала Содействие больным фенилкетонурией в Татарстане
Содействие больным фенилкетонурией в Татарстане Черепно-мозговая травма. Классификация
Черепно-мозговая травма. Классификация Ювенильді ревматоидты артрит
Ювенильді ревматоидты артрит Обмен белков. Переваривание и всасывание. Общие пути обмена
Обмен белков. Переваривание и всасывание. Общие пути обмена Топографическая анатомия нижней конечности. Области мышечной и сосудистой лакуны, бедра, подколенная ямка, ягодичная область
Топографическая анатомия нижней конечности. Области мышечной и сосудистой лакуны, бедра, подколенная ямка, ягодичная область Становление ухода на Руси
Становление ухода на Руси Medical education in Japan
Medical education in Japan Правила снятия ЭКГ. Подготовка пациента к ЭКГ. Какие датчики бывают и как их устанавливать
Правила снятия ЭКГ. Подготовка пациента к ЭКГ. Какие датчики бывают и как их устанавливать Жүкті әйелдің тамақтануы
Жүкті әйелдің тамақтануы Як діють шкідливі звички на організм вагітної жінки
Як діють шкідливі звички на організм вагітної жінки Жеделдеу және созылмалы диссеминирленген туберкулез
Жеделдеу және созылмалы диссеминирленген туберкулез Тромболитики. Механизм действия
Тромболитики. Механизм действия Операции в области живота. Абдоминальная хирургия
Операции в области живота. Абдоминальная хирургия Философия сестринского дела
Философия сестринского дела Первая помощь при утоплении
Первая помощь при утоплении Инфекционная болезнь чума
Инфекционная болезнь чума Цукровий діабет
Цукровий діабет Головокружение и нарушение устойчивости. Современные методы обследования и лечения
Головокружение и нарушение устойчивости. Современные методы обследования и лечения Фармакогнозия как дисциплина
Фармакогнозия как дисциплина