- Хронические лимфопролиферативные заболевания

Содержание
- 2. Хронические лимфопролиферативные заболевания Это опухоли лимфоидной системы, клетки которых могут созревать до морфологически зрелых. Сюда относят
- 3. Хронический лимфолейкоз Наиболее распространенная форма лейкозов в Западном полушарии (20-40% от всех вариантов лейкозов). Чаще у
- 4. Классическая прогрессирующая форма В-ХЛЛ Болеют взрослые, дети никогда, молодые – очень редко. Клиника Увеличены л/у до
- 5. Лабораторные показатели при классической форме ХЛЛ Периферическая кровь: Нв, эритроциты в норме, затем анемия. Развитие анемии
- 6. Лабораторные показатели при классической форме ХЛЛ Костный мозг в большинстве случаев десятки процентов лимфоцитов. Пограничная цифра
- 7. Chronic lymphocyte leukemia CD5+/CD19+ PB(MG) MATURE B-CELL NEOPLASMS
- 8. PB(MG) Hairy cell leukemia -Variant MATURE B-CELL NEOPLASMS
- 9. Т-клеточный вариант ХЛЛ Составляет 3-5% всех случаев ХЛЛ. 3 основные формы: Болезнь Сезари Т-ХЛЛ Грибовидный микоз
- 10. Adult T-cell leukemia /lymphoma MATURE T-CELL NEOPLASMS
- 11. Sezary syndorome:SS Mycosis fungoides:MF Surface antigen Immunophenotyping T cell associated :CD2 95.0% CD5 95.1% CD7 92.9%
- 12. ПАРАПРОТЕИНЕМИЧЕСКИЕ ГЕМОБЛАСТОЗЫ Опухоли системы В-лимфоцитов, дифференцирующихся до стадии секреции моноклонального иммуноглобулина (синонимы: парапротеин, патологический иммуноглобулин).. Нозологические
- 13. Множественная миелома Миеломная болезнь, болезнь Рустицкого-Калера. Эпидемиология: около 1% от всех злокачественных новообразований. Частота встречаемости от
- 14. Патогенез множественной миеломы Пролиферация в КМ (реже в других органах), клона опухолевых В-лимфоцитов. Предполагается опухолевая трансформация
- 15. Классификация миеломной болезни Иммунохимическая классификация (по типу патологического Ig): G-миелома (55—65% случаев) А-миелома (20—25%) D-миелома (2—5%)
- 16. Клиническая картина множественной миеломы Триада симптомов: боли в костях, опухоли, переломы. Боли костях имеют место у
- 17. Клиническая картина множественной миеломы (продолжение) Синдром недостаточности антител. Бактериальные инфекционные осложнения, особенно со стороны легких, дыхательных
- 18. Лабораторные показатели при множественной миеломе Периферическая кровь: Нв, эритроциты могут быть несколько снижены – нормоцитарная анемия
- 19. Лабораторные показатели при множественной миеломе (продолжение) Костный мозг: исследование обязательно для постановки диагноза. Инфильтрация плазматическими клетками
- 20. Диагностические критерии миеломной болезни Критерии первого порядка 30 и более процентов плазматических клеток в КМ парапротеины
- 21. Прогностические критерии МБ Прогноз зависит от стадии заболевания. Основные прогностические показатели: количество и морфология плазматических клеток
- 22. Редкие варианты МБ D-миелома — описано 250 больных. Наблюдается чаще в более молодом возрасте, в основном
- 23. Редкие варианты МБ (продолжение) М-миелома. Описано около 40 наблюдений, среди которых отмечались гепатоспленомегалия, лимфаденопатия, ДВС-синдром, частая
- 24. Клинико-лабораторные особенности миеломы Бенс-Джонса Быстрое развитие почечной недостаточности, гипогамма-глобулинемия, нормальное содержание белка и чаще отсутствие М-градиента
- 25. Солитарная плазмоцитома Возможно это начальная стадия генерализованной плазмоцитомы. Может быть костная и внекостная. Внекостные солитарные плазмоцитомы
- 26. Острый плазмобластный лейкоз. Может быть этапом эволюции МБ (2% случаев МБ трансформируется). Продолжительность жизни менее 1
- 27. Макроглобулинемия Вальденстрема Хронический лейкоз В-клеточной природы, морфологически представленный лимфоцитами, плазмацитами и всеми переходными формами клеток. Частота
- 28. Лабораторные показатели при болезни Вальденстрема Периферическая кровь: Анемия (опухолевое подавление эритропоэза, кровопотеря) Лейкоциты норма (чаще) или
- 29. Болезни тяжелых цепей (БТЦ) В-клеточные опухоли с секрецией фрагментов тяжелых цепей различных классов иммуноглобулинов. БТЦ-γ описана
- 30. Болезни тяжелых цепей (БТЦ) (продолжение) БТЦ-δ (около 200 случаев), у детей, взрослых до 30 лет обоего
- 31. Стадии множественной миеломы по Salmon/Durie I. Нв более 100 г/л, кальций менее 120мг/л, отсутствие остеолиза или
- 32. Стадии множественной миеломы по Salmon/Durie (продолжение) Дополнительно надо оценить массу миеломных клеток по β2М и их
- 33. Диагностика: М-градиент в сыворотке и моче. увеличение кальция в крови. Дифференциальная диагностика: симптоматические парапротеинемии (злокачественные опухоли,
- 35. Скачать презентацию

Хронические лимфопролиферативные заболевания
Это опухоли лимфоидной системы, клетки которых могут созревать до
Хронические лимфопролиферативные заболевания
Это опухоли лимфоидной системы, клетки которых могут созревать до

Хронический лимфолейкоз
Наиболее распространенная форма лейкозов в Западном полушарии (20-40% от всех
Хронический лимфолейкоз
Наиболее распространенная форма лейкозов в Западном полушарии (20-40% от всех

Классическая прогрессирующая форма В-ХЛЛ
Болеют взрослые, дети никогда, молодые – очень редко.
Классическая прогрессирующая форма В-ХЛЛ
Болеют взрослые, дети никогда, молодые – очень редко.

Лабораторные показатели при классической форме ХЛЛ
Периферическая кровь:
Нв, эритроциты в норме,
Лабораторные показатели при классической форме ХЛЛ
Периферическая кровь:
Нв, эритроциты в норме,

Лабораторные показатели при классической форме ХЛЛ
Костный мозг в большинстве случаев
Лабораторные показатели при классической форме ХЛЛ
Костный мозг в большинстве случаев
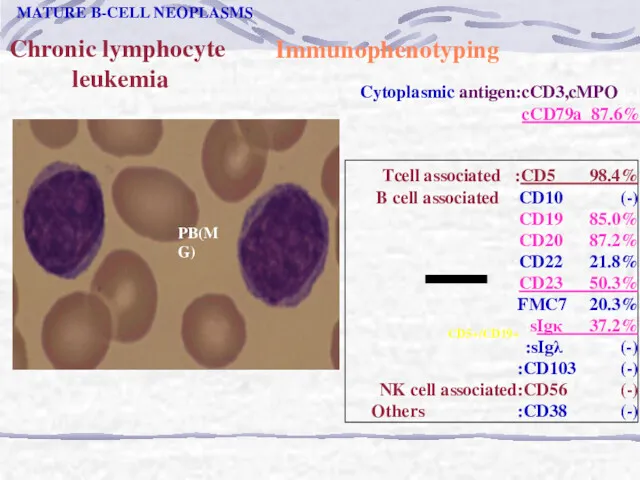
Chronic lymphocyte
leukemia
CD5+/CD19+
PB(MG)
MATURE B-CELL NEOPLASMS
Chronic lymphocyte
leukemia
CD5+/CD19+
PB(MG)
MATURE B-CELL NEOPLASMS
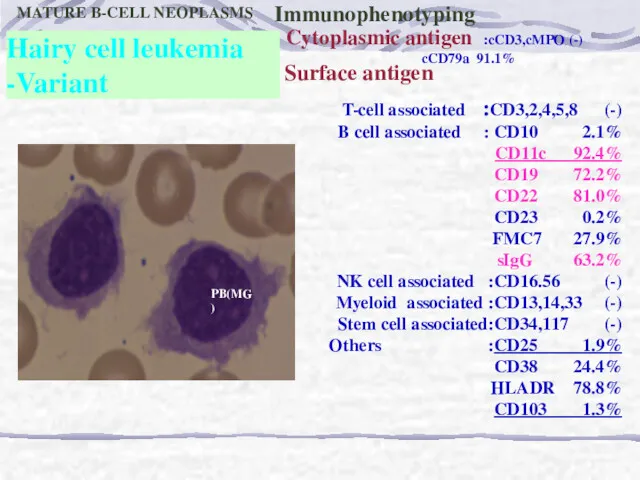
PB(MG)
Hairy cell leukemia
-Variant
MATURE B-CELL NEOPLASMS
PB(MG)
Hairy cell leukemia
-Variant
MATURE B-CELL NEOPLASMS

Т-клеточный вариант ХЛЛ
Составляет 3-5% всех случаев ХЛЛ.
3 основные формы:
Болезнь Сезари
Т-ХЛЛ
Грибовидный
Т-клеточный вариант ХЛЛ
Составляет 3-5% всех случаев ХЛЛ.
3 основные формы:
Болезнь Сезари
Т-ХЛЛ
Грибовидный
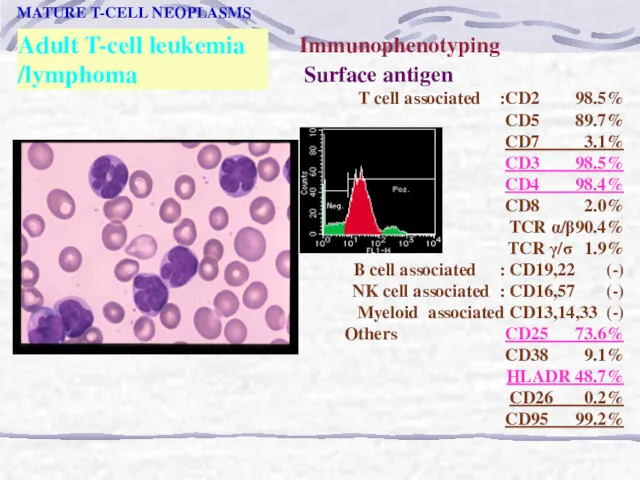
Adult T-cell leukemia
/lymphoma
Adult T-cell leukemia
/lymphoma
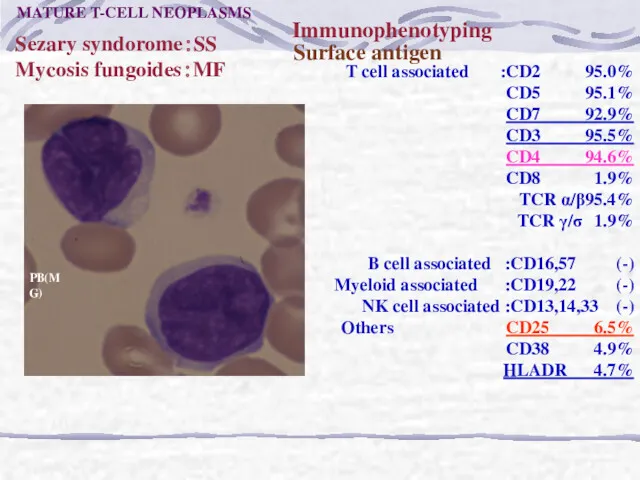
Sezary syndorome:SS
Mycosis fungoides:MF
Surface antigen
Immunophenotyping
T cell associated :CD2 95.0%
CD5
Sezary syndorome:SS
Mycosis fungoides:MF
Surface antigen
Immunophenotyping
T cell associated :CD2 95.0%
CD5

ПАРАПРОТЕИНЕМИЧЕСКИЕ ГЕМОБЛАСТОЗЫ
Опухоли системы В-лимфоцитов, дифференцирующихся до стадии секреции моноклонального иммуноглобулина (синонимы:
ПАРАПРОТЕИНЕМИЧЕСКИЕ ГЕМОБЛАСТОЗЫ
Опухоли системы В-лимфоцитов, дифференцирующихся до стадии секреции моноклонального иммуноглобулина (синонимы:

Множественная миелома
Миеломная болезнь, болезнь Рустицкого-Калера.
Эпидемиология: около 1% от всех злокачественных
Множественная миелома
Миеломная болезнь, болезнь Рустицкого-Калера.
Эпидемиология: около 1% от всех злокачественных

Патогенез множественной миеломы
Пролиферация в КМ (реже в других органах), клона
Патогенез множественной миеломы
Пролиферация в КМ (реже в других органах), клона

Классификация миеломной болезни
Иммунохимическая классификация (по типу патологического Ig):
G-миелома (55—65% случаев)
А-миелома
Классификация миеломной болезни
Иммунохимическая классификация (по типу патологического Ig):
G-миелома (55—65% случаев)
А-миелома

Клиническая картина множественной миеломы
Триада симптомов: боли в костях, опухоли, переломы.
Боли
Клиническая картина множественной миеломы
Триада симптомов: боли в костях, опухоли, переломы.
Боли

Клиническая картина множественной миеломы (продолжение)
Синдром недостаточности антител. Бактериальные инфекционные осложнения, особенно
Клиническая картина множественной миеломы (продолжение)
Синдром недостаточности антител. Бактериальные инфекционные осложнения, особенно

Лабораторные показатели при множественной миеломе
Периферическая кровь:
Нв, эритроциты могут быть
Лабораторные показатели при множественной миеломе
Периферическая кровь:
Нв, эритроциты могут быть

Лабораторные показатели при множественной миеломе (продолжение)
Костный мозг: исследование обязательно для постановки
Лабораторные показатели при множественной миеломе (продолжение)
Костный мозг: исследование обязательно для постановки

Диагностические критерии миеломной болезни
Критерии первого порядка
30 и более процентов плазматических клеток
Диагностические критерии миеломной болезни
Критерии первого порядка
30 и более процентов плазматических клеток

Прогностические критерии МБ
Прогноз зависит от стадии заболевания.
Основные прогностические показатели:
количество
Прогностические критерии МБ
Прогноз зависит от стадии заболевания.
Основные прогностические показатели:
количество

Редкие варианты МБ
D-миелома — описано 250 больных. Наблюдается чаще в более
Редкие варианты МБ
D-миелома — описано 250 больных. Наблюдается чаще в более

Редкие варианты МБ (продолжение)
М-миелома. Описано около 40 наблюдений, среди которых отмечались
Редкие варианты МБ (продолжение)
М-миелома. Описано около 40 наблюдений, среди которых отмечались

Клинико-лабораторные особенности миеломы Бенс-Джонса
Быстрое развитие почечной недостаточности, гипогамма-глобулинемия, нормальное содержание
Клинико-лабораторные особенности миеломы Бенс-Джонса
Быстрое развитие почечной недостаточности, гипогамма-глобулинемия, нормальное содержание

Солитарная плазмоцитома
Возможно это начальная стадия генерализованной плазмоцитомы. Может быть костная
Солитарная плазмоцитома
Возможно это начальная стадия генерализованной плазмоцитомы. Может быть костная

Острый плазмобластный лейкоз.
Может быть этапом эволюции МБ (2% случаев МБ
Острый плазмобластный лейкоз.
Может быть этапом эволюции МБ (2% случаев МБ

Макроглобулинемия Вальденстрема
Хронический лейкоз В-клеточной природы, морфологически представленный лимфоцитами, плазмацитами и
Макроглобулинемия Вальденстрема
Хронический лейкоз В-клеточной природы, морфологически представленный лимфоцитами, плазмацитами и

Лабораторные показатели при болезни Вальденстрема
Периферическая кровь:
Анемия (опухолевое подавление эритропоэза, кровопотеря)
Лейкоциты
Лабораторные показатели при болезни Вальденстрема
Периферическая кровь:
Анемия (опухолевое подавление эритропоэза, кровопотеря)
Лейкоциты

Болезни тяжелых цепей (БТЦ)
В-клеточные опухоли с секрецией фрагментов тяжелых цепей
Болезни тяжелых цепей (БТЦ)
В-клеточные опухоли с секрецией фрагментов тяжелых цепей

Болезни тяжелых цепей (БТЦ) (продолжение)
БТЦ-δ (около 200 случаев), у детей, взрослых
Болезни тяжелых цепей (БТЦ) (продолжение)
БТЦ-δ (около 200 случаев), у детей, взрослых

Стадии множественной миеломы по Salmon/Durie
I. Нв более 100 г/л, кальций менее
Стадии множественной миеломы по Salmon/Durie
I. Нв более 100 г/л, кальций менее

Стадии множественной миеломы по Salmon/Durie (продолжение)
Дополнительно надо оценить массу миеломных клеток
Стадии множественной миеломы по Salmon/Durie (продолжение)
Дополнительно надо оценить массу миеломных клеток

Диагностика: М-градиент в сыворотке и моче. увеличение кальция в крови.
Дифференциальная диагностика:
Диагностика: М-градиент в сыворотке и моче. увеличение кальция в крови.
Дифференциальная диагностика:
 Физика в медицине
Физика в медицине Система крови
Система крови Особенности септического шока у беременных
Особенности септического шока у беременных Нарушение кровообращения. Отеки
Нарушение кровообращения. Отеки Топографическая анатомия и оперативная хирургия печени, желчного пузыря, селезенки и поджелудочной железы
Топографическая анатомия и оперативная хирургия печени, желчного пузыря, селезенки и поджелудочной железы Общее недоразвитие речи. Классификация форм и видов ОНР
Общее недоразвитие речи. Классификация форм и видов ОНР Заболевания крови у детей
Заболевания крови у детей Синдром гострого запалення слизових оболонок дихальних шляхів. Грип
Синдром гострого запалення слизових оболонок дихальних шляхів. Грип Кардиогенный шок
Кардиогенный шок Классические формы ЛВД
Классические формы ЛВД Бүйрек, несепағар, қуықтың даму ауытқулары
Бүйрек, несепағар, қуықтың даму ауытқулары Обсессивно-компульсивное расстройство
Обсессивно-компульсивное расстройство Эволюция представлений о сепсисе. Сепсис-3. Современная концепция патогенеза сепсиса
Эволюция представлений о сепсисе. Сепсис-3. Современная концепция патогенеза сепсиса Классические признаки острого воспаления
Классические признаки острого воспаления Anomalii_refraktsii_dalnozorkost_blizorukost_astigmatizm
Anomalii_refraktsii_dalnozorkost_blizorukost_astigmatizm Бас сүйектерін жасына байланысты оқып білу
Бас сүйектерін жасына байланысты оқып білу Қызыл иектің ультрақұрылысы, қызыл иек сайы, қызыл иек сұйықтығы
Қызыл иектің ультрақұрылысы, қызыл иек сайы, қызыл иек сұйықтығы Ревматические болезни
Ревматические болезни Составление алгоритмов оказания неотложной доврачебной помощи
Составление алгоритмов оказания неотложной доврачебной помощи MythBusters против гомеопатии
MythBusters против гомеопатии Остеохондроз. Этиология
Остеохондроз. Этиология Проводящая система сердца
Проводящая система сердца Артерии. Круги кровообращения. Ангиология
Артерии. Круги кровообращения. Ангиология Заворот толстого отдела кишечника у лошади
Заворот толстого отдела кишечника у лошади Первичный и вторичный иммунный ответ
Первичный и вторичный иммунный ответ Физиология пролактина
Физиология пролактина Диагноз и его обоснование. Дифференциальный диагноз. Планирование лечения
Диагноз и его обоснование. Дифференциальный диагноз. Планирование лечения Укусы насекомых и защита от них
Укусы насекомых и защита от них