- Иммунодефицитные состояния у детей

Содержание
- 2. Первичные иммунодефициты Первичные иммунодефициты (ПИДС) – это врожденные нарушения системы иммунитета, связанные с генетическим дефектом одного
- 3. Актуальность проблемы Первичные иммунодефициты – более частое заболевание, чем это предполагается врачами Частота ПИД может в
- 4. Классификация первичных иммунодефицитов. 1.Комбинированная недостаточность клеточного и гуморального звена иммунитета. Синдромы тяжелого комбинированного иммунодефицита (ТКИД) (20-25
- 5. Особенности инфекций при первичных иммунодефицитах Повторные (чаще обычного) или хронические Мультифокальные Необычно протекающие Необычные возбудители Сопутствующие
- 6. Т-лимфоцитарные/комбинированные иммунодефициты (1:100 000) ТКИД (сцепленный с Х-хромосомой) (50%) Дефицит аденозиндезаминазы (ADA) Синдром голых лимфоцитов Синдром
- 7. Основные возбудители инфекций при Т-лимфоцитарных/комбинированных иммунодефицитах 1.Внутриклеточные бактерии Мycobacteria Listeria Legionella Salmonella Nocardia Chlamydia 2. Грибы
- 8. 3. ДНК-содержащие вирусы herpes simplex virus Varicella zoster virus cytomegalovirus вирус Эпштейна-Барр 4. Простейшие Toxoplasmosis Cryptosporidiosis
- 9. Клинические признаки тяжелого комбинированного иммунодефицита Начало заболевания – первые недели или месяцы жизни Возбудители инфекций -
- 10. Поражение тканей полости рта Candida albicans при ТКИД
- 11. Генерализованный BCGHT (множественные элементы вакцинальной BCG - инфекции в месте вакцинации и на коже тела и
- 12. Лабораторные признаки лимфопения, обусловленная отсутствием Т-лимфоцитов, резкое снижение CD3, CD4, CD8-клеток. резкое снижение функциональной активности Т-лимфоцитов
- 13. Уровень лимфоцитов у детей с различными формами ТКИД
- 14. Синдром Вискотта-Олдрича Частота: 1:250 000 Характер наследования: Х-сцепленный иммунодефицит. Причина: Хp. 11.22-11.23 (WASP) мутация гена, кодирующего
- 15. Синдром Луи-Бар Частота: 1:100000 – 1:1000 000 Характер наследования: аутосомно-рецессивный Причина: IIq 22.3 (atm) – дефект
- 16. Телеангиэктазии на конъюнктиве у пациентки с атаксией-телеангиэктазией
- 17. Синдром Ди-Джорджи Частота: 1:3000 – 1:6000 Причина: дефект в хромосомах 22q11 22q10. Множественные аномалии дериватов 3
- 18. Синдром Ди - Джорджи
- 19. Возможности коррекции Т -лимфоцитарных/комбинированных иммунодефицитов Прогноз при ТКИД – неблагоприятный. Смерть в первые 2 года жизни.
- 20. Недостаточность гуморального звена иммунитета . Болезнь Брутона ( Х-сцепленная агаммаглобулинемия) Общая вариабельная иммунная недостаточность (ОВИН )
- 21. Основные возбудители инфекций при гуморальных иммунодефицитах пиогенные внеклеточные бактерии стрептококки стафилококки пневмококки Hemophilus influensae вирусы энтеровирусы
- 22. Клинические признаки гуморальных иммунодефицитов возраст проявления – 2 полугодие жизни Инфекционные заболевания рецидивирующие синопульмональные инфекции -часто
- 23. Поражение различных органов и систем у больных с гуморальными иммунодефицитами
- 24. Особенности гуморальных иммунодефицитов Болезнь Брутона Частота:1-2 на 100 000 человек Характер наследования: Х-сцепленный Причина: мутация гена,
- 25. . ОВИН Частота: 1:50 000 – 1: 200 000 человек Характер наследования: аутосомно-рецессивный Причина: дефект в
- 26. Множество лямблий (Giardia) на поверхности слизистой оболочки тощей кишки у больного с ОВИН
- 27. Агаммаглобулинемия с повышенным уровнем IgM Частота: во всем мире описано не более 400 больных Характер наследования:
- 28. Пациент с синдромом гипер-IgM, перенесший флегмону орбиты и остеомиелит верхней челюсти
- 29. Селективный дефицит IgA Частота: 1:500 Причина: Молекулярный дефект точно не известен. Блок дифференцировки В-лимфоцитов в IgA-продуцирующие
- 30. Коррекция гуморальных иммунодефицитов противомикробная терапия инфекционных поражений (антибиотики широкого спектра действия в высоких дозах) заместительная терапия
- 31. Дефекты системы фагоцитоза Недостаточность фагоцитов может быть обусловлена нарушением процессов пролиферации, дифференцровки, хемотаксиса нейтрофилов и макрофагов
- 32. Недостаточность системы фагоцитоза Нейтропении (синдром Костманна, циклическая нейтропения) Дефект адгезии лейкоцитов Хроническая гранулематозная болезнь Гипер IgE
- 33. Основные возбудители инфекций при дефектах фагоцитарного звена Грамотрицательные кишечные и пиогенные бактерии: Staphylococcus Pseudomonas Klebsiella E.Coli
- 34. Грибы: Candida Aspergillus sp Mucor mycosis Criptococcus Простейшие: Pneumocystis
- 35. Клинические признаки дефектов фагоцитоза Манифестируют с первых недель, месяцев жизни Эпизоды длительной лихорадки Гнойные инфекции кожи
- 36. Хроническая гранулематозная болезнь Частота: 1:200 000 – 1:500 000 Характер наследования: Х-сцепленный или аутосомно-рецессивный Причина: 1q25.
- 37. Тест восстановления нитросинего тетразолия
- 38. Бактериальная флегмона кожи у ребенка с хронической гранулематозной болезнью
- 39. Регионарный лимфаденит у больного хронической гранулематозной болезнью
- 40. Коррекция дефектов фагоцитоза Профилактические курсы антибактериальной терапии, проивогрибковой терапии Препараты ИФ-γ (усиление хемотаксиса, фагоцитарной и бактерицидной
- 41. Дефицит системы комплемента Дефицит белков комплемента приводит к нарушению выведения из циркулирующей крови микробных тел и
- 42. Клинические проявления Дефицит С1-ингибитора проявляется рецидивирующими отеками Квинке любой локализации Дефицит С1, С2, С4 – развиваются
- 43. Наследственный ангионевротический отек.
- 44. Первичные иммунодефицитные состояния: 10 настораживающих признаков Частые заболевания отитом(не Тяжелые подтвержденные синуситы(не Более двух подтвержденных пневмоний
- 45. Какие анализы должен в первую очередь назначить врач при первичном обследовании больного с подозрением на иммунодефицит
- 46. Регистр первичных иммунодефицитов (56,7%)
- 48. Скачать презентацию

Первичные иммунодефициты
Первичные иммунодефициты (ПИДС) – это врожденные нарушения системы иммунитета,
Первичные иммунодефициты
Первичные иммунодефициты (ПИДС) – это врожденные нарушения системы иммунитета,

Актуальность проблемы
Первичные иммунодефициты – более частое заболевание, чем это предполагается врачами
Частота
Актуальность проблемы
Первичные иммунодефициты – более частое заболевание, чем это предполагается врачами
Частота

Классификация первичных иммунодефицитов.
1.Комбинированная недостаточность клеточного и гуморального звена иммунитета. Синдромы
Классификация первичных иммунодефицитов.
1.Комбинированная недостаточность клеточного и гуморального звена иммунитета. Синдромы

Особенности инфекций при первичных иммунодефицитах
Повторные (чаще обычного) или хронические
Мультифокальные
Необычно
Особенности инфекций при первичных иммунодефицитах
Повторные (чаще обычного) или хронические
Мультифокальные
Необычно

Т-лимфоцитарные/комбинированные иммунодефициты (1:100 000)
ТКИД (сцепленный с Х-хромосомой) (50%)
Дефицит аденозиндезаминазы (ADA)
Синдром голых
Т-лимфоцитарные/комбинированные иммунодефициты (1:100 000)
ТКИД (сцепленный с Х-хромосомой) (50%)
Дефицит аденозиндезаминазы (ADA)
Синдром голых

Основные возбудители инфекций при Т-лимфоцитарных/комбинированных иммунодефицитах
1.Внутриклеточные бактерии
Мycobacteria
Listeria
Основные возбудители инфекций при Т-лимфоцитарных/комбинированных иммунодефицитах
1.Внутриклеточные бактерии
Мycobacteria
Listeria

3. ДНК-содержащие вирусы
herpes simplex virus
Varicella zoster virus
cytomegalovirus вирус
3. ДНК-содержащие вирусы herpes simplex virus Varicella zoster virus cytomegalovirus вирус

Клинические признаки тяжелого комбинированного иммунодефицита
Начало заболевания – первые недели или
Клинические признаки тяжелого комбинированного иммунодефицита
Начало заболевания – первые недели или
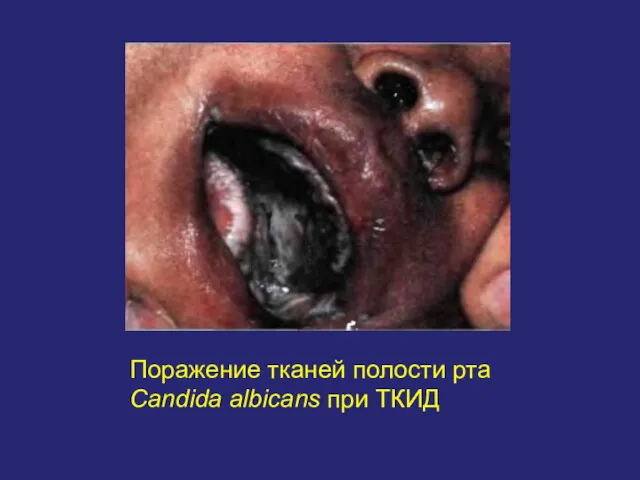
Поражение тканей полости рта Candida albicans при ТКИД
Поражение тканей полости рта Candida albicans при ТКИД
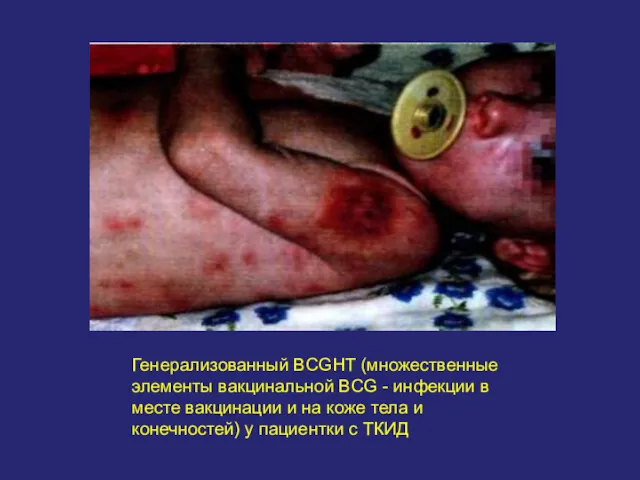
Генерализованный BCGHT (множественные элементы вакцинальной BCG - инфекции в месте вакцинации
Генерализованный BCGHT (множественные элементы вакцинальной BCG - инфекции в месте вакцинации

Лабораторные признаки
лимфопения, обусловленная отсутствием Т-лимфоцитов, резкое снижение CD3, CD4, CD8-клеток.
резкое
Лабораторные признаки
лимфопения, обусловленная отсутствием Т-лимфоцитов, резкое снижение CD3, CD4, CD8-клеток.
резкое
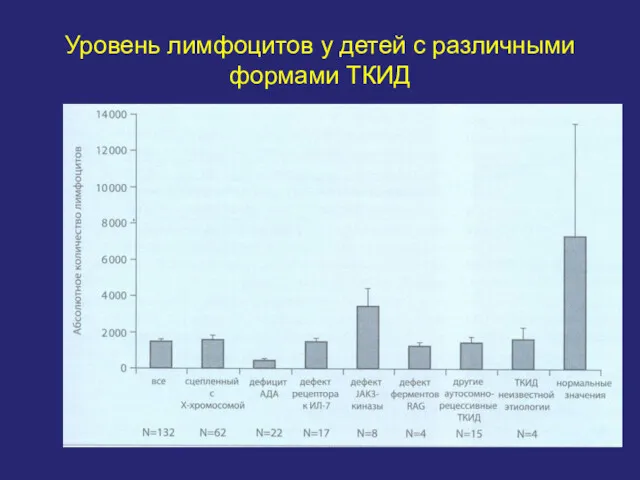
Уровень лимфоцитов у детей с различными формами ТКИД
Уровень лимфоцитов у детей с различными формами ТКИД

Синдром Вискотта-Олдрича
Частота: 1:250 000
Характер наследования: Х-сцепленный иммунодефицит.
Причина: Хp. 11.22-11.23
Синдром Вискотта-Олдрича
Частота: 1:250 000
Характер наследования: Х-сцепленный иммунодефицит.
Причина: Хp. 11.22-11.23

Синдром Луи-Бар
Частота: 1:100000 – 1:1000 000
Характер наследования: аутосомно-рецессивный
Причина: IIq 22.3 (atm)
Синдром Луи-Бар
Частота: 1:100000 – 1:1000 000
Характер наследования: аутосомно-рецессивный
Причина: IIq 22.3 (atm)

Телеангиэктазии на конъюнктиве у пациентки с атаксией-телеангиэктазией
Телеангиэктазии на конъюнктиве у пациентки с атаксией-телеангиэктазией

Синдром Ди-Джорджи
Частота: 1:3000 – 1:6000
Причина: дефект в хромосомах 22q11
Синдром Ди-Джорджи
Частота: 1:3000 – 1:6000
Причина: дефект в хромосомах 22q11

Синдром Ди - Джорджи
Синдром Ди - Джорджи

Возможности коррекции Т -лимфоцитарных/комбинированных иммунодефицитов
Прогноз при ТКИД – неблагоприятный. Смерть в
Возможности коррекции Т -лимфоцитарных/комбинированных иммунодефицитов
Прогноз при ТКИД – неблагоприятный. Смерть в

Недостаточность гуморального звена иммунитета
.
Болезнь Брутона ( Х-сцепленная агаммаглобулинемия)
Общая вариабельная иммунная
Недостаточность гуморального звена иммунитета
.
Болезнь Брутона ( Х-сцепленная агаммаглобулинемия)
Общая вариабельная иммунная

Основные возбудители инфекций при гуморальных иммунодефицитах
пиогенные внеклеточные бактерии
стрептококки
стафилококки
Основные возбудители инфекций при гуморальных иммунодефицитах
пиогенные внеклеточные бактерии
стрептококки
стафилококки

Клинические признаки гуморальных иммунодефицитов
возраст проявления – 2 полугодие жизни
Инфекционные заболевания
рецидивирующие синопульмональные
Клинические признаки гуморальных иммунодефицитов
возраст проявления – 2 полугодие жизни
Инфекционные заболевания
рецидивирующие синопульмональные
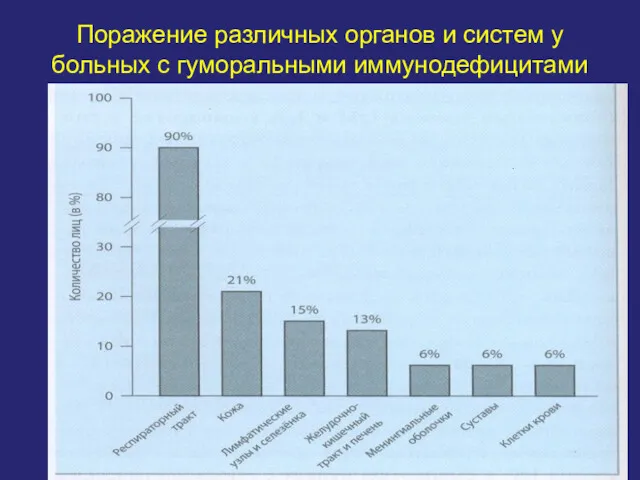
Поражение различных органов и систем у больных с гуморальными иммунодефицитами
Поражение различных органов и систем у больных с гуморальными иммунодефицитами

Особенности гуморальных иммунодефицитов
Болезнь Брутона
Частота:1-2 на 100 000 человек
Характер наследования: Х-сцепленный
Причина:
Особенности гуморальных иммунодефицитов
Болезнь Брутона
Частота:1-2 на 100 000 человек
Характер наследования: Х-сцепленный
Причина:

.
ОВИН
Частота: 1:50 000 – 1: 200 000 человек
Характер наследования: аутосомно-рецессивный
Причина:
.
ОВИН
Частота: 1:50 000 – 1: 200 000 человек
Характер наследования: аутосомно-рецессивный
Причина:
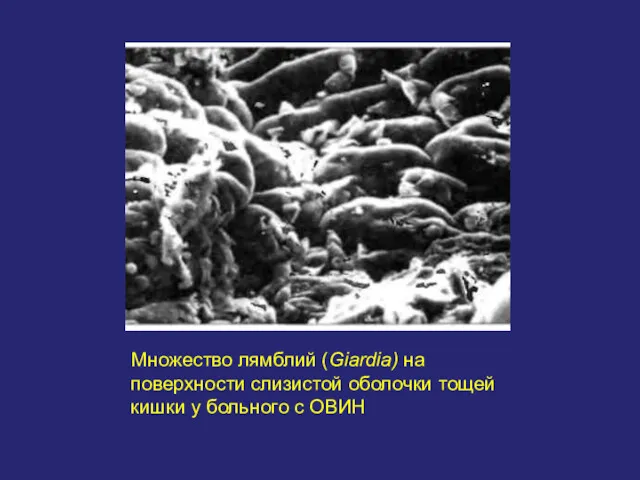
Множество лямблий (Giardia) на поверхности слизистой оболочки тощей кишки у больного
Множество лямблий (Giardia) на поверхности слизистой оболочки тощей кишки у больного

Агаммаглобулинемия с повышенным уровнем IgM
Частота: во всем мире описано
Агаммаглобулинемия с повышенным уровнем IgM
Частота: во всем мире описано
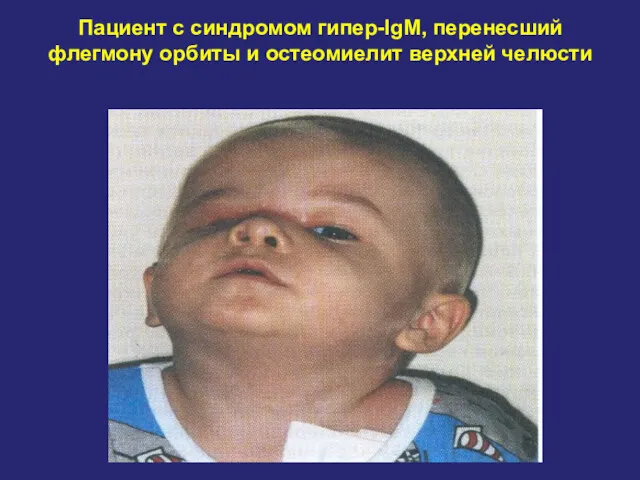
Пациент с синдромом гипер-IgM, перенесший флегмону орбиты и остеомиелит верхней челюсти
Пациент с синдромом гипер-IgM, перенесший флегмону орбиты и остеомиелит верхней челюсти

Селективный дефицит IgA
Частота: 1:500
Причина: Молекулярный дефект точно не известен. Блок
Частота: 1:500
Причина: Молекулярный дефект точно не известен. Блок

Коррекция гуморальных иммунодефицитов
противомикробная терапия инфекционных поражений (антибиотики широкого спектра действия в
Коррекция гуморальных иммунодефицитов
противомикробная терапия инфекционных поражений (антибиотики широкого спектра действия в

Дефекты системы фагоцитоза
Недостаточность фагоцитов может быть обусловлена нарушением процессов пролиферации,
Дефекты системы фагоцитоза
Недостаточность фагоцитов может быть обусловлена нарушением процессов пролиферации,

Недостаточность системы фагоцитоза
Нейтропении (синдром Костманна, циклическая нейтропения)
Дефект адгезии лейкоцитов
Хроническая гранулематозная
Недостаточность системы фагоцитоза
Нейтропении (синдром Костманна, циклическая нейтропения)
Дефект адгезии лейкоцитов
Хроническая гранулематозная

Основные возбудители инфекций при дефектах фагоцитарного звена
Грамотрицательные кишечные и пиогенные бактерии:
Staphylococcus
Основные возбудители инфекций при дефектах фагоцитарного звена
Грамотрицательные кишечные и пиогенные бактерии:
Staphylococcus

Грибы:
Candida
Aspergillus sp
Mucor mycosis
Criptococcus
Простейшие:
Pneumocystis
Грибы:
Candida
Aspergillus sp
Mucor mycosis
Criptococcus
Простейшие:
Pneumocystis

Клинические признаки дефектов фагоцитоза
Манифестируют с первых недель, месяцев жизни
Эпизоды длительной лихорадки
Гнойные
Клинические признаки дефектов фагоцитоза
Манифестируют с первых недель, месяцев жизни
Эпизоды длительной лихорадки
Гнойные

Хроническая гранулематозная болезнь
Частота: 1:200 000 – 1:500 000
Характер наследования: Х-сцепленный или
Хроническая гранулематозная болезнь
Частота: 1:200 000 – 1:500 000
Характер наследования: Х-сцепленный или
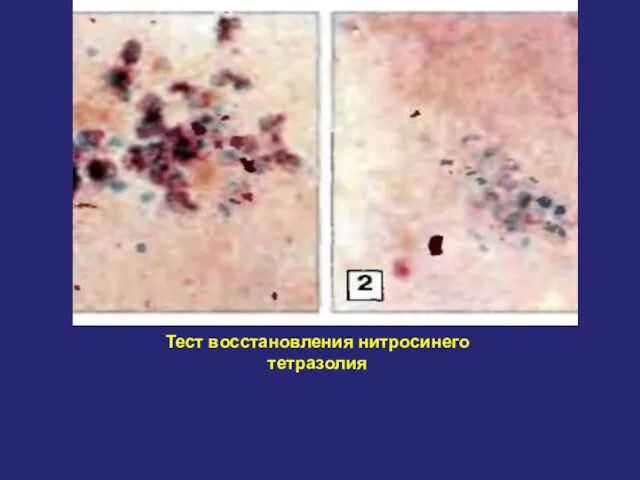
Тест восстановления нитросинего тетразолия
Тест восстановления нитросинего тетразолия
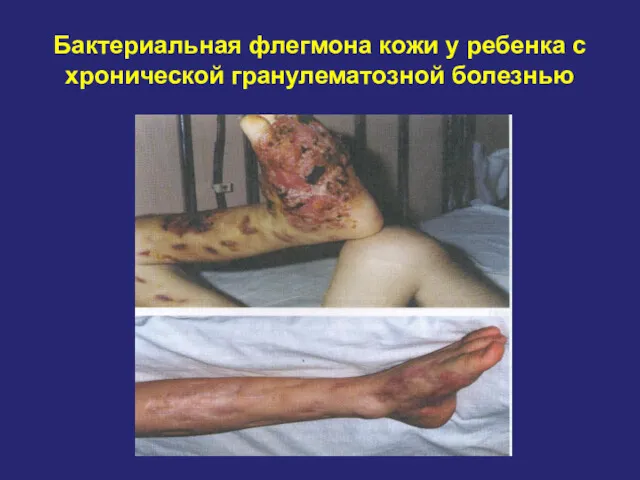
Бактериальная флегмона кожи у ребенка с хронической гранулематозной болезнью
Бактериальная флегмона кожи у ребенка с хронической гранулематозной болезнью
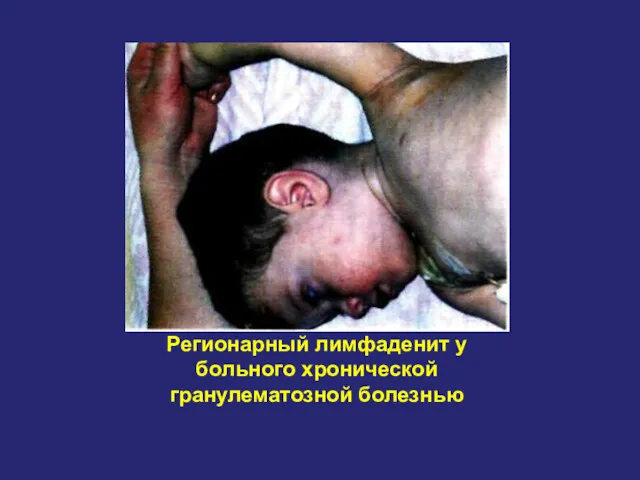
Регионарный лимфаденит у больного хронической гранулематозной болезнью
Регионарный лимфаденит у больного хронической гранулематозной болезнью

Коррекция дефектов фагоцитоза
Профилактические курсы антибактериальной терапии, проивогрибковой терапии
Препараты ИФ-γ (усиление хемотаксиса,
Коррекция дефектов фагоцитоза
Профилактические курсы антибактериальной терапии, проивогрибковой терапии
Препараты ИФ-γ (усиление хемотаксиса,

Дефицит системы комплемента
Дефицит белков комплемента приводит к нарушению выведения из
Дефицит системы комплемента
Дефицит белков комплемента приводит к нарушению выведения из

Клинические проявления
Дефицит С1-ингибитора проявляется рецидивирующими отеками Квинке любой локализации
Дефицит С1, С2,
Клинические проявления
Дефицит С1-ингибитора проявляется рецидивирующими отеками Квинке любой локализации
Дефицит С1, С2,
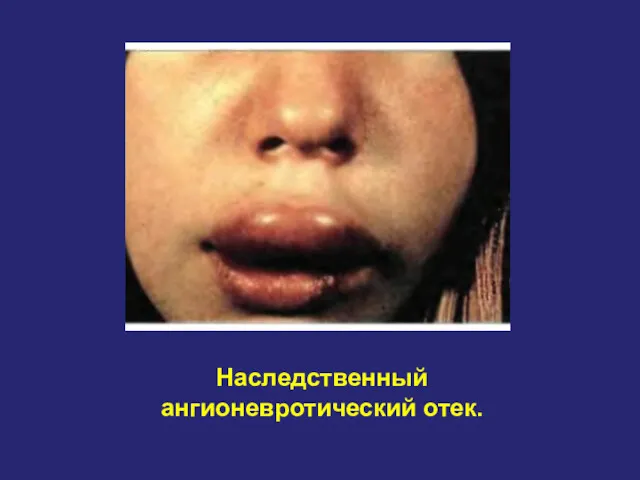
Наследственный ангионевротический отек.
Наследственный ангионевротический отек.

Первичные иммунодефицитные состояния: 10 настораживающих признаков
Частые заболевания отитом(не < 6-8 раз
Первичные иммунодефицитные состояния: 10 настораживающих признаков
Частые заболевания отитом(не < 6-8 раз

Какие анализы должен в первую очередь назначить врач при первичном обследовании
Какие анализы должен в первую очередь назначить врач при первичном обследовании
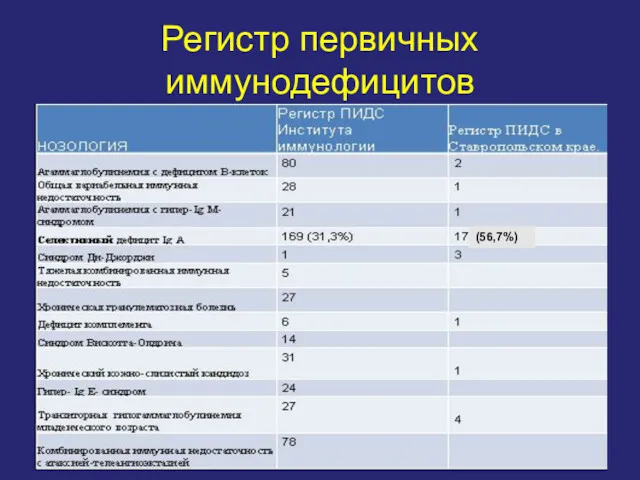
Регистр первичных иммунодефицитов
(56,7%)
Регистр первичных иммунодефицитов
(56,7%)
 Алфлутоп - здоровье суставов в надежных руках
Алфлутоп - здоровье суставов в надежных руках Нейропсихологическая классификация. Критерии выделения форм афазии
Нейропсихологическая классификация. Критерии выделения форм афазии Очаговый туберкулез легких. Туберкулемы. Лекция 7
Очаговый туберкулез легких. Туберкулемы. Лекция 7 Листериоз - Listeria monocytoqenes қоздыратын
Листериоз - Listeria monocytoqenes қоздыратын Анатомо-физиологические особенности подросткового возраста
Анатомо-физиологические особенности подросткового возраста Ложные суставы
Ложные суставы Электрокинетические явления. Коагуляция. Способы очистки коллоидных растворов. Промышленная очистка воды. (Лекция 10)
Электрокинетические явления. Коагуляция. Способы очистки коллоидных растворов. Промышленная очистка воды. (Лекция 10) Инфекциялық иммунология негіздері. Иммунитет түрлері және формалары. Организмнің бейспецификалық қорғаныс
Инфекциялық иммунология негіздері. Иммунитет түрлері және формалары. Организмнің бейспецификалық қорғаныс Туберкулезге қарсы препараттар, фармакокинетикасы, фармакодинамикасы, жанама әсерлері және оларды жою
Туберкулезге қарсы препараттар, фармакокинетикасы, фармакодинамикасы, жанама әсерлері және оларды жою Фермент. Ферменттерді ауруларды диагностикалауға және емдеуге қолдану
Фермент. Ферменттерді ауруларды диагностикалауға және емдеуге қолдану Организм человека и его здоровье ЕГЭ по биологии 2016 год
Организм человека и его здоровье ЕГЭ по биологии 2016 год Диспансеризация. Снижение гинекологических заболевании
Диспансеризация. Снижение гинекологических заболевании Өкпе абцессі
Өкпе абцессі Система автоматизированного анализа степени радиационного поражения человека
Система автоматизированного анализа степени радиационного поражения человека Fruits comment les consommer
Fruits comment les consommer Хирургическое лечение неспецифического язвенного колита и болезни Крона. Неспецифический язвенный колит
Хирургическое лечение неспецифического язвенного колита и болезни Крона. Неспецифический язвенный колит Основы медицинской протозоологии. Тип простейшие. Представители классов споровики и инфузории
Основы медицинской протозоологии. Тип простейшие. Представители классов споровики и инфузории Қызылорда қаласы бойынша №3 қалалық емханада мектепке дейінгі балалардағы вакцина профилактиканы ұйымдастырудағы медбикенің рөлі
Қызылорда қаласы бойынша №3 қалалық емханада мектепке дейінгі балалардағы вакцина профилактиканы ұйымдастырудағы медбикенің рөлі Повреждение и гибель клеток и тканей: причины, механизмы, виды необратимого повреждения. Некроз. Апоптоз
Повреждение и гибель клеток и тканей: причины, механизмы, виды необратимого повреждения. Некроз. Апоптоз Дыхательная гимнастика
Дыхательная гимнастика Pneumonia. Currently, several types of pneumonia are distinguished
Pneumonia. Currently, several types of pneumonia are distinguished Қоғамдық денсаулық ғылым және оқыту пәні ретінде
Қоғамдық денсаулық ғылым және оқыту пәні ретінде Blood pressure. Measurement
Blood pressure. Measurement Әртүрлі жүйедегі бекітілістерді қолдана отырып алмалы конструкцияларды әзірлеу технологиясы
Әртүрлі жүйедегі бекітілістерді қолдана отырып алмалы конструкцияларды әзірлеу технологиясы Кизилунгач, ошкозон – ичак касалликлари
Кизилунгач, ошкозон – ичак касалликлари Основы эпидемиологии. (Лекция 5)
Основы эпидемиологии. (Лекция 5) Нарушения мышления: расстройства ассоциативного процесса. Патология суждений и умозаключений
Нарушения мышления: расстройства ассоциативного процесса. Патология суждений и умозаключений Економіка охорони здоров'я як наука і практика. Ціноутворення медичних послуг у стоматології
Економіка охорони здоров'я як наука і практика. Ціноутворення медичних послуг у стоматології