- Иммунодефициты. Определение иммунодефицита

Содержание
- 2. Лекция 1 Иммунодефициты
- 3. Определение иммунодефицита – под иммунодефицитами понимают состояния, характеризующиеся неспособностью организма развивать полноценную иммунную реакцию на патоген
- 4. Классификация первичных иммунодефицитов Комбинированные иммунодефициты Т-клеточные иммунодефициты Гуморальные иммунодефициты Дефициты системы фагоцитов Дефицит системы комплемента
- 5. Комбинированные иммунодефициты Классификация Тяжелый комбинированный иммунодефицит с ретикулярной дигенезией (ретикулярный дисгенез) Тяжелый комбинированный иммунодефицит - Х-сцепленный
- 6. Комбинированные иммунодефициты Уровни нарушений в системе иммуногенеза. При комбинированных иммунодефицитах нарушается процесс формирования из стволовых кроветворных
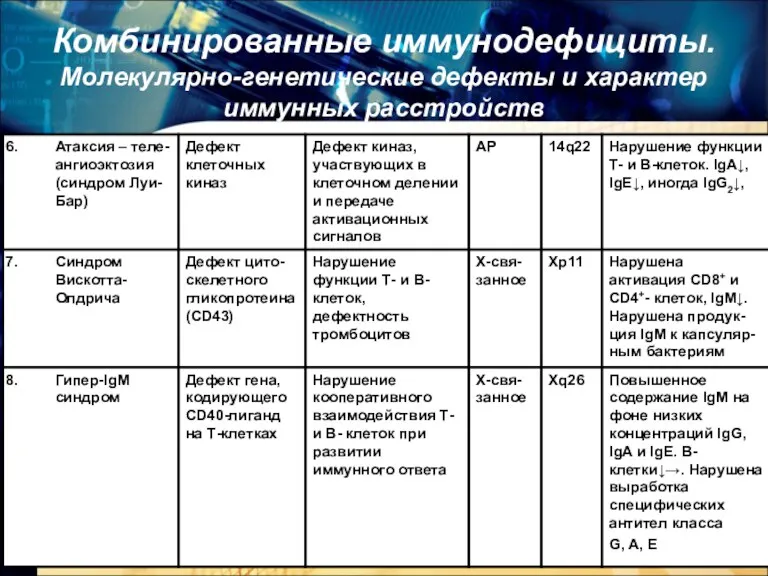
- 7. Комбинированные иммунодефициты. Молекулярно-генетические дефекты и характер иммунных расстройств
- 8. Комбинированные иммунодефициты. Молекулярно-генетические дефекты и характер иммунных расстройств
- 9. Комбинированные иммунодефициты. Молекулярно-генетические дефекты и характер иммунных расстройств
- 10. Комбинированные иммунодефициты Общим для комбинированных иммунодефицитов является: Раннее их появление ( в возрасте 2-7 мес.) Инфицирование
- 11. Комбинированные иммунодефициты Клиническая характеристика Дети с ПКИД страдают от тяжело протекающих бактериальных, вирусных и грибковых инфекций
- 12. Комбинированные иммунодефициты Для ПКИД характерно: Лимфоцитопения Гипоплазия тимуса Гипоплазия лимфатических узлов, миндалин Снижение количества Т-клеток в
- 13. Комбинированные иммунодефициты 1.ТКИД с ретикулярной дисгенезией (ретикулярный дисгенез) Для заболевания характерно: В костном мозге практически отсутствуют
- 14. Комбинированные иммунодефициты 2.ТКИД: Х-сцепленный тип (болеют только мальчики) Аутосомно-рецессивный тип (болеют в равной степени мальчики и
- 15. Комбинированные иммунодефициты Мальчик с первичной тяжелой комбинированной иммунной недостаточностью. У малыша выраженная дистрофия, вздутый живот, вертикальные
- 16. Комбинированные иммунодефициты 3.Иммунодефицит, сцепленный с дефицитом АДА (аденозиндезаминазой) Заболевание связано с дефицитом аденозиндезаминазы в лимфоцитах. Ее
- 17. Комбинированные иммунодефициты 4. Иммунодефицит, сцепленный с дефицитом ПНФ (пуриннуклеозидфосфорилазы) Развитие синдрома связано с недостаточностью в лимфоцитах
- 18. Комбинированные иммунодефициты 4. Иммунодефицит, сцепленный с дефицитом ПНФ (пуриннуклеозидфосфорилазы) В некоторых случаях развиваются аутоиммунные расстройства Аутоиммунная
- 19. Комбинированные иммунодефициты 5. Синдром «голых» лимфоцитов (дефицит экспрессии на лимфоцитах НLA-антигенов I и II класса) Синдром
- 20. Комбинированные иммунодефициты 5. Синдром «голых» лимфоцитов (дефицит экспрессии на лимфоцитах НLA-антигенов I и II класса) Дети
- 21. Комбинированные иммунодефициты 6. Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар) Заболевание связано с дефективностью киназ, участвующих в регуляции
- 22. Комбинированные иммунодефициты 6. Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар) У больных наблюдается триада: ИД + сосудистая патология
- 23. Комбинированные иммунодефициты 6. Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар) Проявления: Кожно-глазные телеангиэктазии (телеангиэктазии мелких сосудов расположенных в
- 24. Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар) Иммунные нарушения: Снижено количество Т-лимфоцитов Количество В-лимфоцитов, как правило, в пределах
- 25. Комбинированные иммунодефициты 7. Синдром Вискотта-Олдрича Заболевание развивается только у мальчиков (Х-сцепленный тип наследования). Манифест с 6-месячного
- 26. Синдром Вискотта-Олдрича Иммунные нарушения: снижено количество Т-лимфоцитов в крови, количество В-лимфоцитов – в норме снижено содержание
- 27. Комбинированные иммунодефициты 8. Синдром Незелофа У детей с этим синдромом наблюдается недоразвитый эмбриоподобный тимус, который не
- 28. Синдром Незелофа Лечение Трансплантация тимуса Препараты тимуса Препараты иммуноглобулинов Противомикробные средства
- 29. Т-клеточные иммунодефициты Классификация Т-клеточных иммунодефицитов Синдром Ди-Джорджи Хронический кожно-слизистый кандидоз
- 30. Т-клеточные иммунодефициты Специфические дефекты и характер иммунных расстройств у детей с первичными Т-лимфоцитарными иммунодефицитами.
- 31. Т-клеточные иммунодефициты Уровни нарушений в системе иммуногенеза
- 32. Т-клеточные иммунодефициты 1.Синдром Ди-Джорджи Для заболевания характерна триада аномалий: Гипоплазия тимуса Гипоплазия паращитовидных желез Аномалия дуги
- 33. Т-клеточные иммунодефициты 1.Синдром Ди-Джорджи Кардиальные нарушения проявляются: цианозом одышкой в покое шумовой симптоматикой при аускультации Недоразвитие
- 34. Т-клеточные иммунодефициты 2. Хронический кожно-слизистый кандидоз Клинически Т-клеточный кандидоз проявляется в поражении кожи, ногтей, волосистой части
- 35. Дефицит гуморального иммунитета (антителообразования) Классификация Наследственная гипогаммаглобулинемия, сцепленная с Х-хромосомой (болезнь Брутона) Несемейная гипогаммаглобулинемия (D80.1). Общий
- 36. Дефицит гуморального иммунитета (антителообразования) Общим для этой группы ИД является: Заболевания возникает со второго полугодия жизни
- 37. Дефицит гуморального иммунитета (антителообразования) Молекулярно-генетические дефекты и характер иммунных расстройств у детей с первичными гуморальными иммунодефицитами
- 38. Дефицит гуморального иммунитета (антителообразования) Уровни нарушения в системе иммуногенеза при гуморальном иммунодефиците
- 39. Дефицит гуморального иммунитета Наследственная гипогаммаглобулинемия, сцепленная с Х-хромосомой (болезнь Брутона) Заболевание сцеплено с Х-хромосомой, болеют только
- 40. Дефицит гуморального иммунитета 1. Болезнь Брутона Клинически заболевание манифестирует хроническими рецидивирующими инфекциями дыхательной системы, ЛОР-органов, кожи,
- 41. Дефицит гуморального иммунитета Болезнь Брутона Лечение: Заместительная терапия ВВИГ (нормальный человеческий иммуноглобулин для внутривенного введения). Заместительная
- 42. Дефицит гуморального иммунитета 2. Несемейная гипогаммаглобулинемия (D80.1) Болеют мальчики и девочки. По клинике и иммунным нарушениям
- 43. Дефицит гуморального иммунитета 3. Общий вариабельный иммунодефицит (общая вариабельная гипогаммаглобулинемия) Заболевание может возникнуть в любом возрасте,
- 44. Дефицит гуморального иммунитета 3. Общий вариабельный иммунодефицит (общая вариабельная гипогаммаглобулинемия) Лечение: Антимикробная терапия Заместительная терапия антителосодержащими
- 45. Дефицит гуморального иммунитета 4. Транзиторная гипогаммаглобулинемия детей (медленный иммунологический ответ) Естественное иммунодефицитное состояние, встречающееся у 5-8%
- 46. Дефицит гуморального иммунитета 4. Транзиторная гипогаммаглобулинемия детей (медленный иммунологический ответ) Лечение: Симптоматическое, направленное на купирование инфекций
- 47. Дефицит гуморального иммунитета 5. Дисгаммаглобулинемии (избирательный дефицит отдельных классов иммуноглобулинов) Клинически заболевания манифестируют патологией, характерной для
- 48. Дефицит гуморального иммунитета Различают следующие разновидности дисгаммаглобулинемий: 5.1 Селективный дефицит IgA Ставится детям старше 1 года,
- 49. Дефицит гуморального иммунитета 5. Дисгаммаглобулинемии Лечение: Симптоматическое, подавление инфекции (антибиотики) Иммуностимуляция препаратами (рибомунил, бронхомунил, биостим, ликопид,
- 50. Дефицит гуморального иммунитета 6. Гипер-IgM синдром Х-сцепленное заболевание. Клинически заболевание манифестирует патологией, характерной для группы гуморальных
- 51. Дефицит гуморального иммунитета 6. Гипер-IgM синдром Лечение: Антимикробная терапия Препараты ВВИГ (вначале в режиме насыщения, затем
- 52. Дефицит системы фагоцитов Дефицит системы фагоцитов манифестирует Хроническим гранулематозом Синдромом Чедиака-Хигаси Синдромом Джоба (гипериммуноглобулинемия Е) Дефицитом
- 53. Дефицит системы фагоцитов Молекулярно-генетические дефекты и характер иммунных расстройств у больных с первичным дефицитом системы фагоцитов
- 54. Дефицит системы фагоцитов Молекулярно-генетические дефекты и характер иммунных расстройств у больных с первичным дефицитом системы фагоцитов
- 55. Дефицит системы фагоцитов
- 56. Дефицит системы фагоцитов Хронический гранулематоз Заболевание может появиться в детстве, так и у взрослых. Возникновение заболевания
- 57. Дефицит системы фагоцитов Мальчик 9 лет с хронической стафилококковой инфекцией, связанной с недостаточностью внутриклеточного переваривания бактерий
- 58. Дефицит системы фагоцитов Хронический гранулематоз Иммунологические расстройства ПМЯЛ, макрофаги способны к киллингу и перевариванию поглощенных микробов
- 59. Дефицит системы фагоцитов Хронический гранулематоз Лечение: Антибактериальная и противогрибковая терапия – пожизненно (терапия проводится даже в
- 60. Дефицит системы фагоцитов 2. Синдром Чедиака-Хигаси Развитие инфекции связано с потерей способности нейтрофилов переваривать микробы (нарушена
- 61. Дефицит системы фагоцитов 2. Синдром Чедиака-Хигаси Лечение: Антимикробные средства Симптоматическое лечение Высокие дозы витамина С (500
- 62. Дефицит системы фагоцитов 3. Синдром гипериммуноглобулинемии (синдром Джоба) Клинические проявления: атипичное течение воспаления ( без повышения
- 63. Дефицит системы фагоцитов 3. Синдром Джоба Лабораторные показатели: высокая концентрация в крови IgE (1000 МЕ/мл) высокая
- 64. Дефицит системы фагоцитов 4. Дефицит экспрессии молекул адгезии (дефицит адгезии лейкоцитов) Клинически заболевание проявляется инфекционным поражением
- 65. Дефицит системы комплемента Различают следующие виды: Дефицит С1, С4, С2 Дефицит С3 Дефицит С5-С9 Дефицит С3b-ингибитора
- 66. Дефицит системы комплемента Клинические проявления дефицита компонента комплемента
- 67. Дефицит системы комплемента Наследственный ангионевротический отек Клинические проявления: Рецидивирующие самоорганизующиеся отеки кожи и слизистых покровов (глотки,
- 68. Дефицит системы комплемента Наследственный ангионевротический отек Провоцирующие факторы Травма Операции Психоэмоциональные и физические перенапряжения Химические и
- 69. Дефицит системы комплемента Наследственный ангионевротический отек Лечение: В острый период: Переливание нативной или свежезамороженной плазмы, не
- 70. Дефицит системы комплемента Наследственный ангионевротический отек Лечение: При наличии противопоказаний для назначения андрогенов их заменяют на
- 72. Скачать презентацию

Лекция 1
Иммунодефициты
Лекция 1
Иммунодефициты
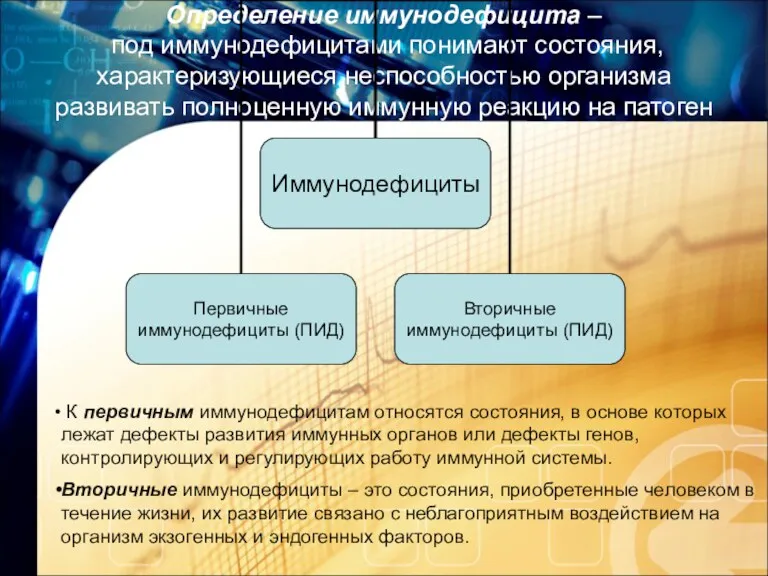
Определение иммунодефицита –
под иммунодефицитами понимают состояния, характеризующиеся неспособностью организма развивать
Определение иммунодефицита – под иммунодефицитами понимают состояния, характеризующиеся неспособностью организма развивать

Классификация
первичных иммунодефицитов
Комбинированные иммунодефициты
Т-клеточные иммунодефициты
Гуморальные иммунодефициты
Дефициты системы фагоцитов
Дефицит системы комплемента
Классификация
первичных иммунодефицитов
Комбинированные иммунодефициты
Т-клеточные иммунодефициты
Гуморальные иммунодефициты
Дефициты системы фагоцитов
Дефицит системы комплемента

Комбинированные иммунодефициты
Классификация
Тяжелый комбинированный иммунодефицит с ретикулярной дигенезией (ретикулярный дисгенез)
Тяжелый комбинированный
Комбинированные иммунодефициты
Классификация
Тяжелый комбинированный иммунодефицит с ретикулярной дигенезией (ретикулярный дисгенез)
Тяжелый комбинированный
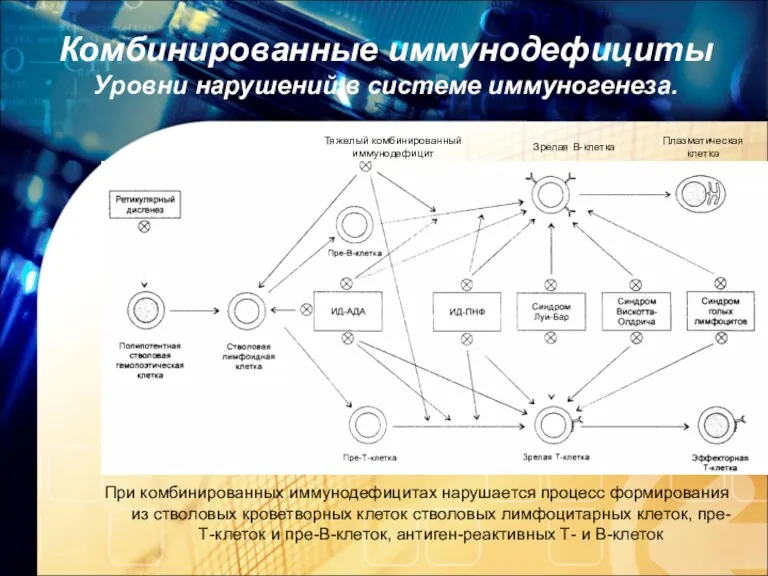
Комбинированные иммунодефициты Уровни нарушений в системе иммуногенеза.
При комбинированных иммунодефицитах нарушается процесс
Комбинированные иммунодефициты Уровни нарушений в системе иммуногенеза.
При комбинированных иммунодефицитах нарушается процесс
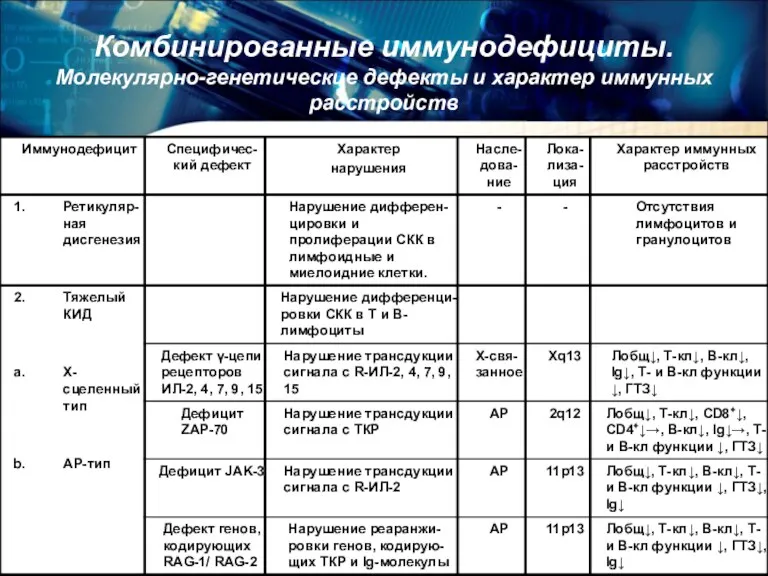
Комбинированные иммунодефициты.
Молекулярно-генетические дефекты и характер иммунных расстройств
Комбинированные иммунодефициты.
Молекулярно-генетические дефекты и характер иммунных расстройств
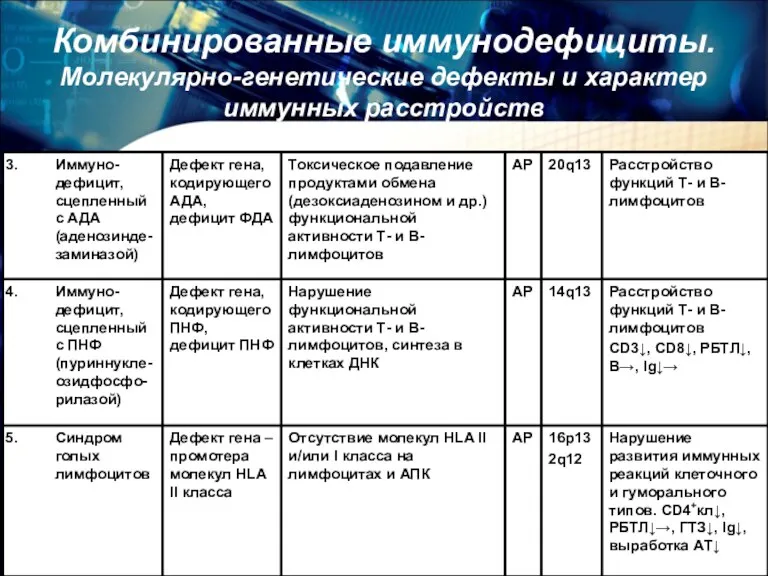
Комбинированные иммунодефициты.
Молекулярно-генетические дефекты и характер иммунных расстройств
Комбинированные иммунодефициты.
Молекулярно-генетические дефекты и характер иммунных расстройств
Комбинированные иммунодефициты.
Молекулярно-генетические дефекты и характер иммунных расстройств
Комбинированные иммунодефициты.
Молекулярно-генетические дефекты и характер иммунных расстройств

Комбинированные иммунодефициты
Общим для комбинированных иммунодефицитов является:
Раннее их появление ( в возрасте
Комбинированные иммунодефициты
Общим для комбинированных иммунодефицитов является:
Раннее их появление ( в возрасте

Комбинированные иммунодефициты
Клиническая характеристика
Дети с ПКИД страдают от тяжело протекающих бактериальных,
Комбинированные иммунодефициты
Клиническая характеристика
Дети с ПКИД страдают от тяжело протекающих бактериальных,

Комбинированные иммунодефициты
Для ПКИД характерно:
Лимфоцитопения
Гипоплазия тимуса
Гипоплазия лимфатических узлов, миндалин
Снижение количества Т-клеток в
Комбинированные иммунодефициты
Для ПКИД характерно:
Лимфоцитопения
Гипоплазия тимуса
Гипоплазия лимфатических узлов, миндалин
Снижение количества Т-клеток в

Комбинированные иммунодефициты
1.ТКИД с ретикулярной дисгенезией
(ретикулярный дисгенез)
Для заболевания характерно:
В костном мозге практически
Комбинированные иммунодефициты
1.ТКИД с ретикулярной дисгенезией
(ретикулярный дисгенез)
Для заболевания характерно:
В костном мозге практически

Комбинированные иммунодефициты
2.ТКИД:
Х-сцепленный тип (болеют только мальчики)
Аутосомно-рецессивный тип (болеют в равной степени
Комбинированные иммунодефициты
2.ТКИД:
Х-сцепленный тип (болеют только мальчики)
Аутосомно-рецессивный тип (болеют в равной степени
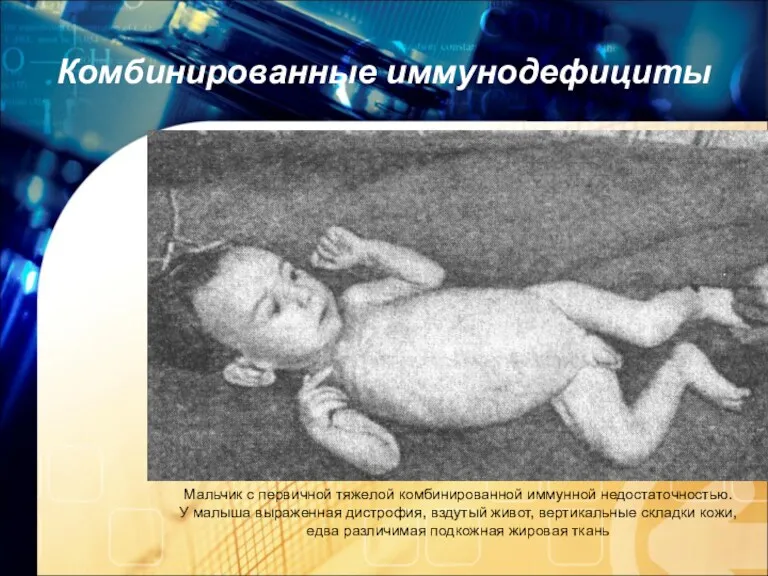
Комбинированные иммунодефициты
Мальчик с первичной тяжелой комбинированной иммунной недостаточностью.
У малыша выраженная дистрофия,
Комбинированные иммунодефициты
Мальчик с первичной тяжелой комбинированной иммунной недостаточностью.
У малыша выраженная дистрофия,

Комбинированные иммунодефициты
3.Иммунодефицит, сцепленный с дефицитом АДА (аденозиндезаминазой)
Заболевание связано с дефицитом аденозиндезаминазы
Комбинированные иммунодефициты
3.Иммунодефицит, сцепленный с дефицитом АДА (аденозиндезаминазой)
Заболевание связано с дефицитом аденозиндезаминазы

Комбинированные иммунодефициты
4. Иммунодефицит, сцепленный с дефицитом ПНФ (пуриннуклеозидфосфорилазы)
Развитие синдрома связано с
Комбинированные иммунодефициты
4. Иммунодефицит, сцепленный с дефицитом ПНФ (пуриннуклеозидфосфорилазы)
Развитие синдрома связано с

Комбинированные иммунодефициты
4. Иммунодефицит, сцепленный с дефицитом ПНФ (пуриннуклеозидфосфорилазы)
В некоторых случаях развиваются
Комбинированные иммунодефициты
4. Иммунодефицит, сцепленный с дефицитом ПНФ (пуриннуклеозидфосфорилазы)
В некоторых случаях развиваются

Комбинированные иммунодефициты
5. Синдром «голых» лимфоцитов (дефицит экспрессии на лимфоцитах НLA-антигенов I
Комбинированные иммунодефициты
5. Синдром «голых» лимфоцитов (дефицит экспрессии на лимфоцитах НLA-антигенов I

Комбинированные иммунодефициты
5. Синдром «голых» лимфоцитов (дефицит экспрессии на лимфоцитах НLA-антигенов I
Комбинированные иммунодефициты
5. Синдром «голых» лимфоцитов (дефицит экспрессии на лимфоцитах НLA-антигенов I

Комбинированные иммунодефициты
6. Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар)
Заболевание связано с дефективностью киназ,
Комбинированные иммунодефициты
6. Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар)
Заболевание связано с дефективностью киназ,

Комбинированные иммунодефициты
6. Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар)
У больных наблюдается триада:
ИД
Комбинированные иммунодефициты
6. Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар)
У больных наблюдается триада:
ИД
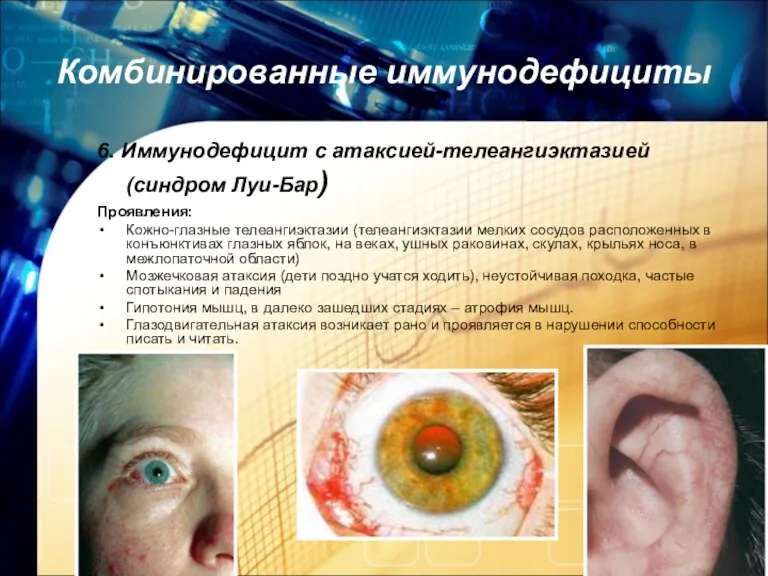
Комбинированные иммунодефициты
6. Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар)
Проявления:
Кожно-глазные телеангиэктазии (телеангиэктазии мелких
Комбинированные иммунодефициты
6. Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар)
Проявления:
Кожно-глазные телеангиэктазии (телеангиэктазии мелких

Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар)
Иммунные нарушения:
Снижено количество Т-лимфоцитов
Количество В-лимфоцитов,
Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар)
Иммунные нарушения:
Снижено количество Т-лимфоцитов
Количество В-лимфоцитов,

Комбинированные иммунодефициты
7. Синдром Вискотта-Олдрича
Заболевание развивается только у мальчиков (Х-сцепленный тип наследования).
Комбинированные иммунодефициты
7. Синдром Вискотта-Олдрича
Заболевание развивается только у мальчиков (Х-сцепленный тип наследования).
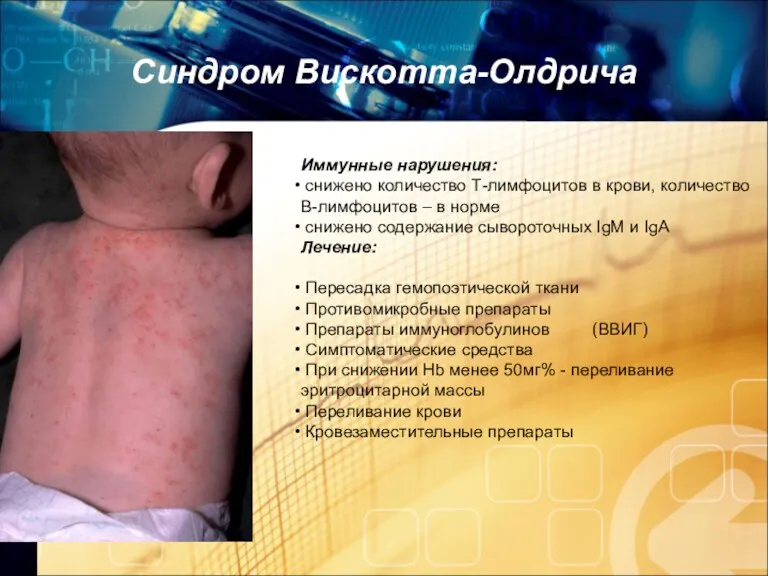
Синдром Вискотта-Олдрича
Иммунные нарушения:
снижено количество Т-лимфоцитов в крови, количество В-лимфоцитов
Синдром Вискотта-Олдрича
Иммунные нарушения:
снижено количество Т-лимфоцитов в крови, количество В-лимфоцитов

Комбинированные иммунодефициты
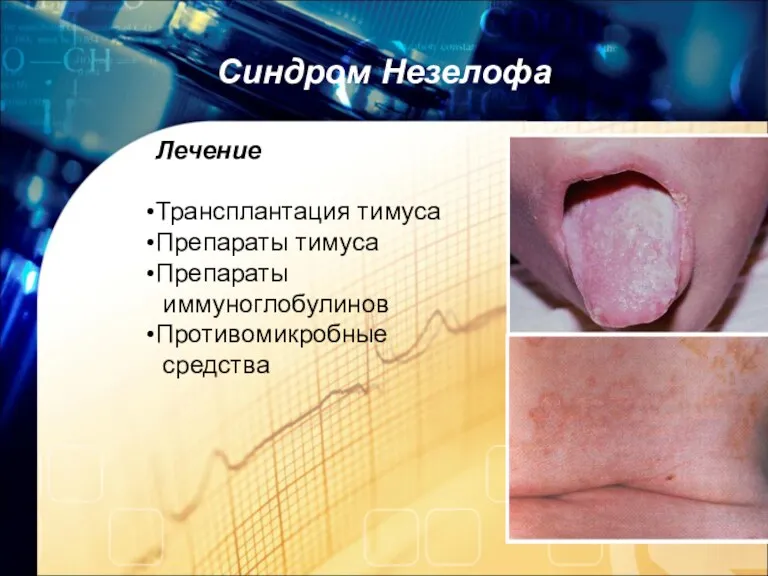
8. Синдром Незелофа
У детей с этим синдромом наблюдается недоразвитый
Комбинированные иммунодефициты
8. Синдром Незелофа
У детей с этим синдромом наблюдается недоразвитый
Синдром Незелофа
Лечение
Трансплантация тимуса
Препараты тимуса
Препараты
иммуноглобулинов
Противомикробные
средства
Синдром Незелофа
Лечение
Трансплантация тимуса
Препараты тимуса
Препараты
иммуноглобулинов
Противомикробные
средства

Т-клеточные иммунодефициты
Классификация Т-клеточных иммунодефицитов
Синдром Ди-Джорджи
Хронический кожно-слизистый кандидоз
Т-клеточные иммунодефициты
Классификация Т-клеточных иммунодефицитов
Синдром Ди-Джорджи
Хронический кожно-слизистый кандидоз
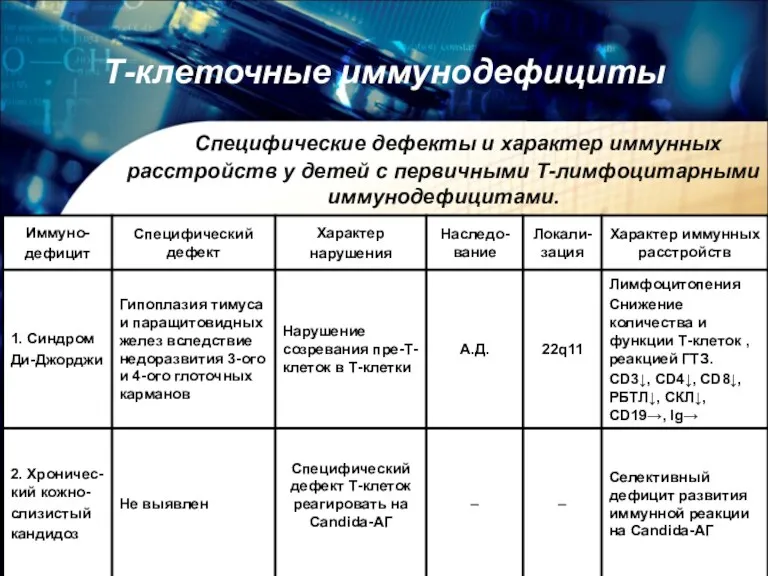
Т-клеточные иммунодефициты
Специфические дефекты и характер иммунных расстройств у детей с
Т-клеточные иммунодефициты
Специфические дефекты и характер иммунных расстройств у детей с
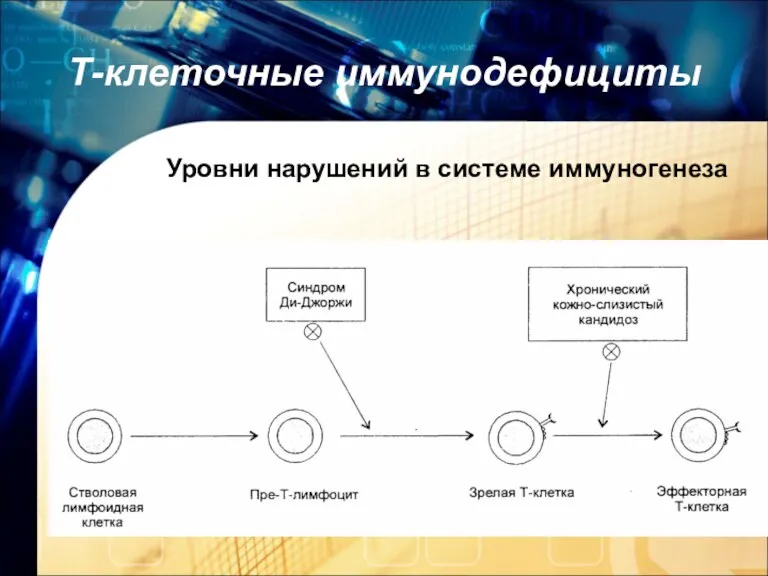
Т-клеточные иммунодефициты
Уровни нарушений в системе иммуногенеза
Т-клеточные иммунодефициты
Уровни нарушений в системе иммуногенеза

Т-клеточные иммунодефициты
1.Синдром Ди-Джорджи
Для заболевания характерна триада аномалий:
Гипоплазия тимуса
Гипоплазия паращитовидных желез
Аномалия
Т-клеточные иммунодефициты
1.Синдром Ди-Джорджи
Для заболевания характерна триада аномалий:
Гипоплазия тимуса
Гипоплазия паращитовидных желез
Аномалия

Т-клеточные иммунодефициты
1.Синдром Ди-Джорджи
Кардиальные нарушения проявляются:
цианозом
одышкой в покое
шумовой
Т-клеточные иммунодефициты
1.Синдром Ди-Джорджи
Кардиальные нарушения проявляются:
цианозом
одышкой в покое
шумовой

Т-клеточные иммунодефициты
2. Хронический кожно-слизистый кандидоз
Клинически Т-клеточный кандидоз проявляется в поражении
Т-клеточные иммунодефициты
2. Хронический кожно-слизистый кандидоз
Клинически Т-клеточный кандидоз проявляется в поражении

Дефицит гуморального иммунитета (антителообразования)
Классификация
Наследственная гипогаммаглобулинемия, сцепленная с
Х-хромосомой (болезнь Брутона)
Несемейная
Дефицит гуморального иммунитета (антителообразования)
Классификация
Наследственная гипогаммаглобулинемия, сцепленная с
Х-хромосомой (болезнь Брутона)
Несемейная

Дефицит гуморального иммунитета (антителообразования)
Общим для этой группы ИД является:
Заболевания возникает со
Дефицит гуморального иммунитета (антителообразования)
Общим для этой группы ИД является:
Заболевания возникает со
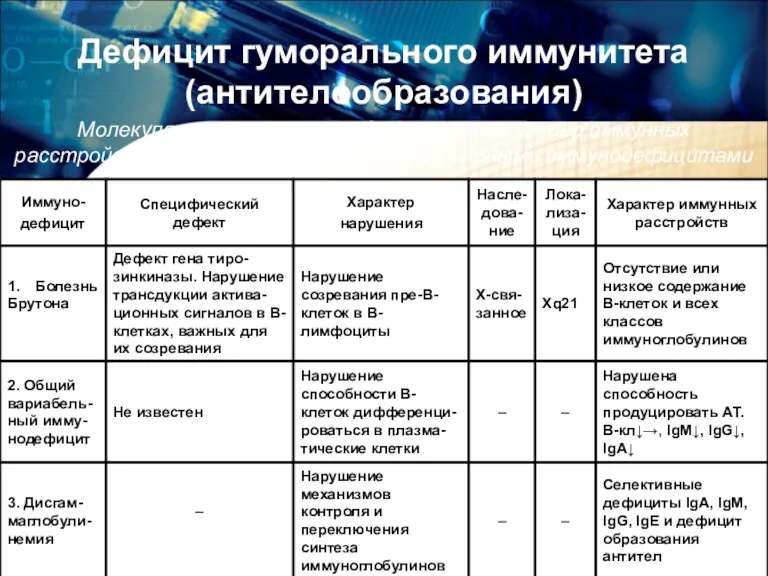
Дефицит гуморального иммунитета (антителообразования)
Молекулярно-генетические дефекты и характер иммунных расстройств у детей
Дефицит гуморального иммунитета (антителообразования)
Молекулярно-генетические дефекты и характер иммунных расстройств у детей
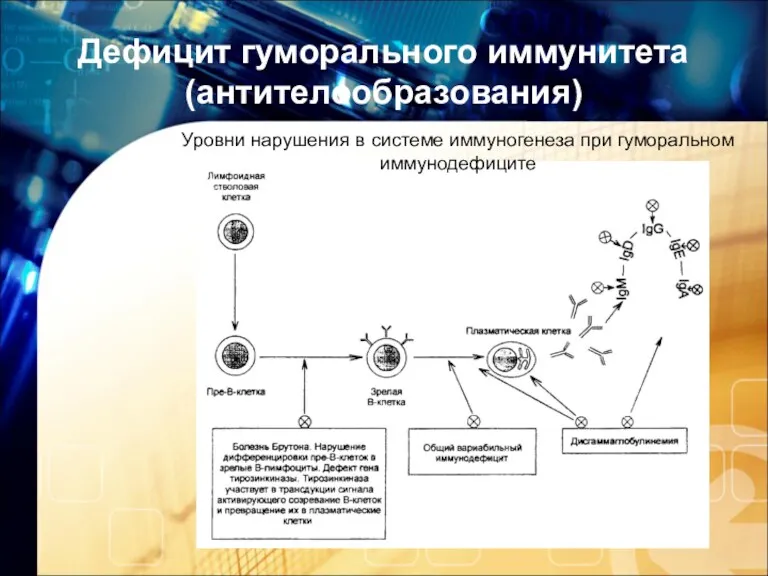
Дефицит гуморального иммунитета (антителообразования)
Уровни нарушения в системе иммуногенеза при гуморальном иммунодефиците
Дефицит гуморального иммунитета (антителообразования)
Уровни нарушения в системе иммуногенеза при гуморальном иммунодефиците

Дефицит гуморального иммунитета
Наследственная гипогаммаглобулинемия, сцепленная с Х-хромосомой (болезнь Брутона)
Заболевание
Дефицит гуморального иммунитета
Наследственная гипогаммаглобулинемия, сцепленная с Х-хромосомой (болезнь Брутона)
Заболевание

Дефицит гуморального иммунитета
1. Болезнь Брутона
Клинически заболевание манифестирует хроническими рецидивирующими
Дефицит гуморального иммунитета
1. Болезнь Брутона
Клинически заболевание манифестирует хроническими рецидивирующими

Дефицит гуморального иммунитета
Болезнь Брутона
Лечение:
Заместительная терапия ВВИГ (нормальный человеческий иммуноглобулин для
Дефицит гуморального иммунитета
Болезнь Брутона
Лечение:
Заместительная терапия ВВИГ (нормальный человеческий иммуноглобулин для

Дефицит гуморального иммунитета
2. Несемейная гипогаммаглобулинемия (D80.1)
Болеют мальчики и девочки.
По
Дефицит гуморального иммунитета
2. Несемейная гипогаммаглобулинемия (D80.1)
Болеют мальчики и девочки.
По

Дефицит гуморального иммунитета
3. Общий вариабельный иммунодефицит (общая вариабельная гипогаммаглобулинемия)
Заболевание может возникнуть
Дефицит гуморального иммунитета
3. Общий вариабельный иммунодефицит (общая вариабельная гипогаммаглобулинемия)
Заболевание может возникнуть

Дефицит гуморального иммунитета
3. Общий вариабельный иммунодефицит (общая вариабельная гипогаммаглобулинемия)
Лечение:
Антимикробная терапия
Заместительная терапия
Дефицит гуморального иммунитета
3. Общий вариабельный иммунодефицит (общая вариабельная гипогаммаглобулинемия)
Лечение:
Антимикробная терапия
Заместительная терапия

Дефицит гуморального иммунитета
4. Транзиторная гипогаммаглобулинемия детей (медленный иммунологический ответ)
Естественное иммунодефицитное состояние,
Дефицит гуморального иммунитета
4. Транзиторная гипогаммаглобулинемия детей (медленный иммунологический ответ)
Естественное иммунодефицитное состояние,

Дефицит гуморального иммунитета
4. Транзиторная гипогаммаглобулинемия детей (медленный иммунологический ответ)
Лечение:
Симптоматическое, направленное на
Дефицит гуморального иммунитета
4. Транзиторная гипогаммаглобулинемия детей (медленный иммунологический ответ)
Лечение:
Симптоматическое, направленное на

Дефицит гуморального иммунитета
5. Дисгаммаглобулинемии
(избирательный дефицит отдельных классов иммуноглобулинов)
Клинически заболевания манифестируют
Дефицит гуморального иммунитета
5. Дисгаммаглобулинемии
(избирательный дефицит отдельных классов иммуноглобулинов)
Клинически заболевания манифестируют

Дефицит гуморального иммунитета
Различают следующие разновидности дисгаммаглобулинемий:
5.1 Селективный дефицит IgA
Ставится детям
Дефицит гуморального иммунитета
Различают следующие разновидности дисгаммаглобулинемий:
5.1 Селективный дефицит IgA
Ставится детям

Дефицит гуморального иммунитета
5. Дисгаммаглобулинемии
Лечение:
Симптоматическое, подавление инфекции (антибиотики)
Иммуностимуляция препаратами (рибомунил, бронхомунил, биостим,
Дефицит гуморального иммунитета
5. Дисгаммаглобулинемии
Лечение:
Симптоматическое, подавление инфекции (антибиотики)
Иммуностимуляция препаратами (рибомунил, бронхомунил, биостим,

Дефицит гуморального иммунитета
6. Гипер-IgM синдром
Х-сцепленное заболевание.
Клинически заболевание манифестирует патологией,
Дефицит гуморального иммунитета
6. Гипер-IgM синдром
Х-сцепленное заболевание.
Клинически заболевание манифестирует патологией,

Дефицит гуморального иммунитета
6. Гипер-IgM синдром
Лечение:
Антимикробная терапия
Препараты ВВИГ (вначале в режиме насыщения,
Дефицит гуморального иммунитета
6. Гипер-IgM синдром
Лечение:
Антимикробная терапия
Препараты ВВИГ (вначале в режиме насыщения,
Дефицит системы фагоцитов
Дефицит системы фагоцитов манифестирует
Хроническим гранулематозом
Синдромом Чедиака-Хигаси
Синдромом Джоба (гипериммуноглобулинемия Е)
Дефицитом
Дефицит системы фагоцитов
Дефицит системы фагоцитов манифестирует
Хроническим гранулематозом
Синдромом Чедиака-Хигаси
Синдромом Джоба (гипериммуноглобулинемия Е)
Дефицитом
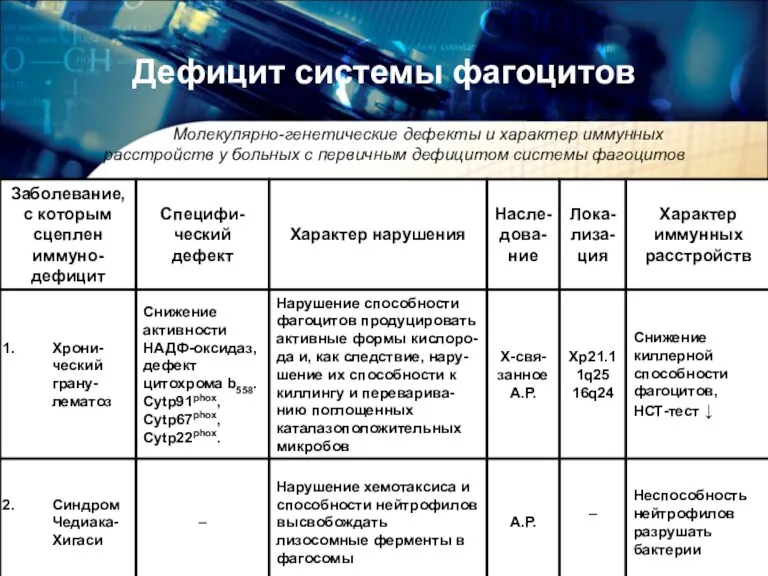
Дефицит системы фагоцитов
Молекулярно-генетические дефекты и характер иммунных расстройств у больных
Дефицит системы фагоцитов
Молекулярно-генетические дефекты и характер иммунных расстройств у больных

Дефицит системы фагоцитов
Молекулярно-генетические дефекты и характер иммунных расстройств у больных с
Дефицит системы фагоцитов
Молекулярно-генетические дефекты и характер иммунных расстройств у больных с
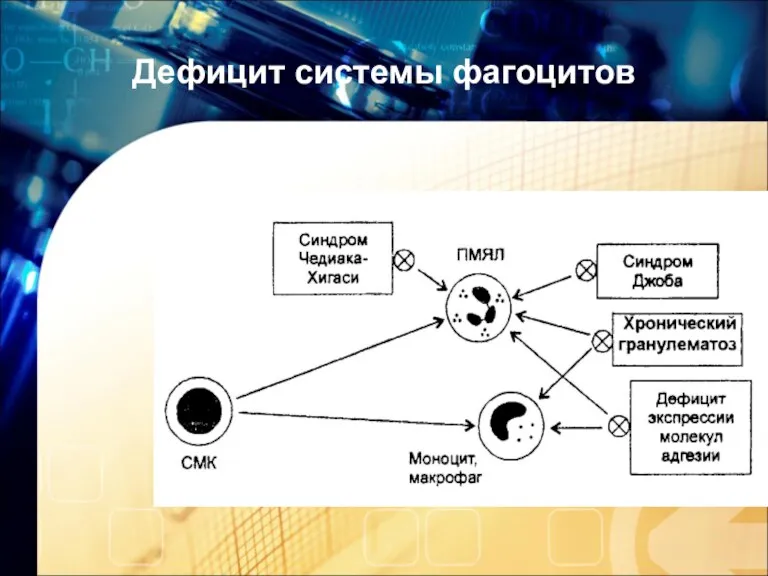
Дефицит системы фагоцитов
Дефицит системы фагоцитов

Дефицит системы фагоцитов
Хронический гранулематоз
Заболевание может появиться в детстве, так и у
Дефицит системы фагоцитов
Хронический гранулематоз
Заболевание может появиться в детстве, так и у
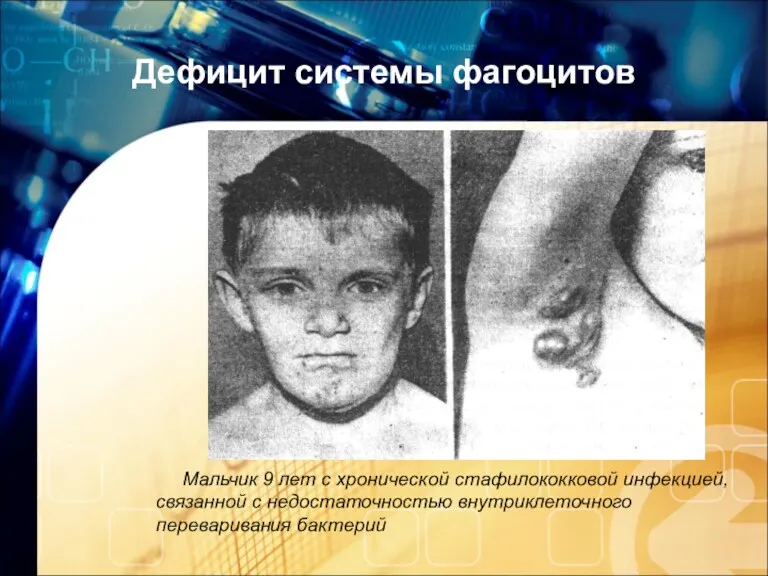
Дефицит системы фагоцитов
Мальчик 9 лет с хронической стафилококковой инфекцией, связанной
Дефицит системы фагоцитов
Мальчик 9 лет с хронической стафилококковой инфекцией, связанной

Дефицит системы фагоцитов
Хронический гранулематоз
Иммунологические расстройства
ПМЯЛ, макрофаги способны к киллингу и перевариванию
Дефицит системы фагоцитов
Хронический гранулематоз
Иммунологические расстройства
ПМЯЛ, макрофаги способны к киллингу и перевариванию

Дефицит системы фагоцитов
Хронический гранулематоз
Лечение:
Антибактериальная и противогрибковая терапия – пожизненно (терапия
Дефицит системы фагоцитов
Хронический гранулематоз
Лечение:
Антибактериальная и противогрибковая терапия – пожизненно (терапия

Дефицит системы фагоцитов
2. Синдром Чедиака-Хигаси
Развитие инфекции связано с потерей способности
Дефицит системы фагоцитов
2. Синдром Чедиака-Хигаси
Развитие инфекции связано с потерей способности

Дефицит системы фагоцитов
2. Синдром Чедиака-Хигаси
Лечение:
Антимикробные средства
Симптоматическое лечение
Высокие дозы витамина С
Дефицит системы фагоцитов
2. Синдром Чедиака-Хигаси
Лечение:
Антимикробные средства
Симптоматическое лечение
Высокие дозы витамина С

Дефицит системы фагоцитов
3. Синдром гипериммуноглобулинемии (синдром Джоба)
Клинические проявления:
атипичное течение воспаления (
Дефицит системы фагоцитов
3. Синдром гипериммуноглобулинемии (синдром Джоба)
Клинические проявления:
атипичное течение воспаления (

Дефицит системы фагоцитов
3. Синдром Джоба
Лабораторные показатели:
высокая концентрация в крови IgE (1000
Дефицит системы фагоцитов
3. Синдром Джоба
Лабораторные показатели:
высокая концентрация в крови IgE (1000

Дефицит системы фагоцитов
4. Дефицит экспрессии молекул адгезии
(дефицит адгезии лейкоцитов)
Клинически
Дефицит системы фагоцитов
4. Дефицит экспрессии молекул адгезии
(дефицит адгезии лейкоцитов)
Клинически

Дефицит системы комплемента
Различают следующие виды:
Дефицит С1, С4, С2
Дефицит С3
Дефицит С5-С9
Дефицит С3b-ингибитора
Дефицит системы комплемента
Различают следующие виды:
Дефицит С1, С4, С2
Дефицит С3
Дефицит С5-С9
Дефицит С3b-ингибитора

Дефицит системы комплемента
Клинические проявления дефицита компонента комплемента
Дефицит системы комплемента
Клинические проявления дефицита компонента комплемента

Дефицит системы комплемента
Наследственный ангионевротический отек
Клинические проявления:
Рецидивирующие самоорганизующиеся отеки кожи и
Дефицит системы комплемента
Наследственный ангионевротический отек
Клинические проявления:
Рецидивирующие самоорганизующиеся отеки кожи и

Дефицит системы комплемента
Наследственный ангионевротический отек
Провоцирующие факторы
Травма
Операции
Психоэмоциональные и физические перенапряжения
Химические и лекарственные
Дефицит системы комплемента
Наследственный ангионевротический отек
Провоцирующие факторы
Травма
Операции
Психоэмоциональные и физические перенапряжения
Химические и лекарственные

Дефицит системы комплемента
Наследственный ангионевротический отек
Лечение:
В острый период:
Переливание нативной
Дефицит системы комплемента
Наследственный ангионевротический отек
Лечение:
В острый период:
Переливание нативной

Дефицит системы комплемента
Наследственный ангионевротический отек
Лечение:
При наличии противопоказаний для назначения андрогенов
Дефицит системы комплемента
Наследственный ангионевротический отек
Лечение:
При наличии противопоказаний для назначения андрогенов
 Переломы костей верхних конечностей. Клиника, диагностика, дифференциальная диагностика. Оказание первой помощи и лечение
Переломы костей верхних конечностей. Клиника, диагностика, дифференциальная диагностика. Оказание первой помощи и лечение Антиатеросклеротические (гиполипидемические и эндотелиотропные) средства
Антиатеросклеротические (гиполипидемические и эндотелиотропные) средства Грыжи передней брюшной стенки
Грыжи передней брюшной стенки Жүрек ырғағы мен өткізгіштігінің жіті бұзылыстары. Кенеттен болған өлім
Жүрек ырғағы мен өткізгіштігінің жіті бұзылыстары. Кенеттен болған өлім Афазия
Афазия Реализация регионального проекта Укрепление общественного здоровья
Реализация регионального проекта Укрепление общественного здоровья Асфиксия и рН пуповинной крови
Асфиксия и рН пуповинной крови Фотоконкурс Если хочешь быть здоров
Фотоконкурс Если хочешь быть здоров Медико-санитарное обеспечение при ликвидации последствий радиационных аварий
Медико-санитарное обеспечение при ликвидации последствий радиационных аварий Носовые кровотечения. (Лекция 3)
Носовые кровотечения. (Лекция 3) Деформация зубных рядов. Зубочелюстная аномалия
Деформация зубных рядов. Зубочелюстная аномалия Функциональная магнитно-резонансная томография
Функциональная магнитно-резонансная томография Характеристика популярных диет
Характеристика популярных диет Бронхиальная астма. Лечение обострений, неотложная помощь
Бронхиальная астма. Лечение обострений, неотложная помощь Клинический разбор
Клинический разбор Всероссийская служба медицины катастроф. Основы лечебно-эвакуационного обеспечения населения в ЧС мирного и военного времени
Всероссийская служба медицины катастроф. Основы лечебно-эвакуационного обеспечения населения в ЧС мирного и военного времени 1 декабря – Всемирный день борьбы со СПИДом
1 декабря – Всемирный день борьбы со СПИДом Неспецифический язвенный колит и болезнь Крона
Неспецифический язвенный колит и болезнь Крона Первая помощь при ДТП
Первая помощь при ДТП Утомление, переутомление, перетренированность. Хроническое физическое перенапряжение систем организма. Внезапная смерть в спорте
Утомление, переутомление, перетренированность. Хроническое физическое перенапряжение систем организма. Внезапная смерть в спорте Грипп и другие ОРВИ
Грипп и другие ОРВИ Антитела, строение и функции
Антитела, строение и функции Гігієна харчування, або трофогігієна
Гігієна харчування, або трофогігієна Функциональный анализ зубочелюстной системы. Элементы окклюзионной поверхности зубов и зубных рядов
Функциональный анализ зубочелюстной системы. Элементы окклюзионной поверхности зубов и зубных рядов Көздің ісіктері
Көздің ісіктері Қанның қан тамырларымен қозғалысының гемодинамикалық заңдылықтары. (Дәріс 11)
Қанның қан тамырларымен қозғалысының гемодинамикалық заңдылықтары. (Дәріс 11) Қант диабетінің І-типі және IIтипінің диагностикасы
Қант диабетінің І-типі және IIтипінің диагностикасы Dental faculty
Dental faculty