- Классификация иммунодефицитных состояний

Содержание
- 2. Классификация иммунодефицитных состояний В зависимости от уровня дефекта: Т-клеточный иммунодефицит В-клеточный иммунодефицит Дефект неспецифической резистентности (фагоцитарных
- 3. Первичные иммунодефициты 1.Связанные с дефектами фагоцитов 2.Дефицит системы комплемента 3. Т-клеточные иммунодефициты 4. В-клеточные иммунодефициты 5.Тяжелые
- 4. Дефицит системы фагоцитов 10-12% от общей частоты встречаемости перв.ИД Хронический гранулематоз (в большинстве случаев связано с
- 5. Клинические проявление хронического гранулематоза Рецидивирующие бактериальные инфекции Образование в тканях гранулем Проявляется, как правило, в детском
- 6. Синдром Чегиак-Хигаши (Чедиак-Хигаси) Наследуется по аутосомно-рецессивному типу. Потеря нейтрофилами способности высвобождать лизосомальные ферменты (нарушена структура и
- 7. Клинические проявления Расстройство пигментации : общая гипопигментация и пигментная дистрофия (светлая прозрачная кожа, светлые редкие сухие
- 8. Дефицит системы комплемента 1% от общего количества перв.ИД Генетические дефекты описаны для всех компонентов комплемента С1q,
- 9. Синдром Ди Джорджи Аплазия (полный Ди Джорджи) или гипоплазия тимуса (частичный Ди Джорджи) Триада -недоразвитие тимуса
- 10. Синдром Ди Джорджи Иммунологические последствия Снижение количества и активности Т-лф Количество В-лф в норме Уровень ат
- 11. Синдром Ди Джорджи Клиника: Рецидивирующие вирусные, паразитарные, вирусные инфекции Судороги Дисморфия лица Другие пороки развития –атрезия
- 12. Синдром Незелофа Гипоплазия тимуса Может наследоваться по аутосомно-рецессивному пути и быть сцеплена с полом клиника: замедление
- 13. Синдром Незелофа
- 14. Дефицит В-клеточного звена Агамма(гипогамма)глобулинемия (синдром Брутона) рецессивный тип наследования, встречается в основном у мальчиков Дефицит цитоплазматической
- 15. Синдром Брутона Снижено количество иммуноглобулинов всех классов, но в большей степени IgG Отсутствуют плазматические клетки в
- 16. клиника Определяется на 5-9 месяце жизни Рецидивирующие гнойные инфекции Менингиты Отсутствие реакции лф.узлов и селезенки на
- 17. Дефицит IgA Различают недостаточность полную или селективную (IgA2) Развиваются частые респираторные инфекции Заместительная терапия не показана
- 18. Х-сцепленная агаммаглобулинемия с гиперглобулинемией М Развитие связано с невозможностью переключения синтеза иммуноглобулинов с IgM на другие
- 19. Х-сцепленная агаммаглобулинемия с гиперглобулинемией М Лабораторные данные: IgA, IgG,IgE – не определяются Уровень IgM значительно повышен,
- 20. ТКИД (швейцарский тип) Х-сцепленный тип Дефект на уровне стволовой клетки нежизнеспособность
- 21. Комбинированные ИД Синдром Вискотта-Олдрича Нарушение активации Т-h и Т-s ) Врожденный дефект тромбоцитов Нарушение способности макрофагов
- 22. Синдром Вискотта-Олдрича Клиника: Экзема Тромбоцитопения Бактериальные и вирусные инфекции
- 23. Синдром Вискотта-Олдрича
- 24. Синдром Луи-Бар (атаксия-телеангиэктазия) Гипоплазия тимуса Гипоплазия лимф. узлов, селезенки Количественная и функциональная недостаточность Т-лимфоцитов Снижение количества
- 25. Синдром Луи-Бар Клиника: Атаксия Телеангиэктазы (на носу, ушах, конъюктиве) Рецидивирующие инфекции Опухолевые заболевания
- 27. Скачать презентацию

Классификация иммунодефицитных состояний
В зависимости от уровня дефекта:
Т-клеточный иммунодефицит
В-клеточный иммунодефицит
Дефект неспецифической резистентности
(фагоцитарных
Классификация иммунодефицитных состояний
В зависимости от уровня дефекта:
Т-клеточный иммунодефицит
В-клеточный иммунодефицит
Дефект неспецифической резистентности
(фагоцитарных

Первичные иммунодефициты
1.Связанные с дефектами фагоцитов
2.Дефицит системы комплемента
3. Т-клеточные иммунодефициты
4. В-клеточные
Первичные иммунодефициты
1.Связанные с дефектами фагоцитов
2.Дефицит системы комплемента
3. Т-клеточные иммунодефициты
4. В-клеточные

Дефицит системы фагоцитов
10-12% от общей частоты встречаемости перв.ИД
Хронический гранулематоз
(в
Дефицит системы фагоцитов
10-12% от общей частоты встречаемости перв.ИД
Хронический гранулематоз
(в

Клинические проявление хронического гранулематоза
Рецидивирующие бактериальные инфекции
Образование в тканях гранулем
Проявляется, как правило,
Клинические проявление хронического гранулематоза
Рецидивирующие бактериальные инфекции
Образование в тканях гранулем
Проявляется, как правило,

Синдром Чегиак-Хигаши
(Чедиак-Хигаси)
Наследуется по аутосомно-рецессивному типу.
Потеря нейтрофилами способности высвобождать лизосомальные ферменты
Синдром Чегиак-Хигаши
(Чедиак-Хигаси)
Наследуется по аутосомно-рецессивному типу.
Потеря нейтрофилами способности высвобождать лизосомальные ферменты

Клинические проявления
Расстройство пигментации : общая гипопигментация и пигментная дистрофия (светлая прозрачная кожа,
Клинические проявления
Расстройство пигментации : общая гипопигментация и пигментная дистрофия (светлая прозрачная кожа,

Дефицит системы комплемента
1% от общего количества перв.ИД
Генетические дефекты описаны для всех
Дефицит системы комплемента
1% от общего количества перв.ИД
Генетические дефекты описаны для всех

Синдром Ди Джорджи
Аплазия (полный Ди Джорджи) или гипоплазия тимуса (частичный
Синдром Ди Джорджи
Аплазия (полный Ди Джорджи) или гипоплазия тимуса (частичный
Синдром Ди Джорджи
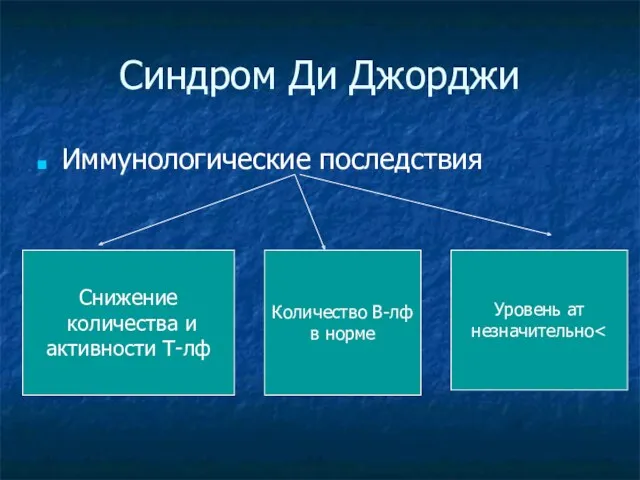
Иммунологические последствия
Снижение
количества и
активности Т-лф
Количество В-лф
в норме
Уровень
Синдром Ди Джорджи
Иммунологические последствия
Снижение
количества и
активности Т-лф
Количество В-лф
в норме
Уровень

Синдром
Ди Джорджи
Клиника:
Рецидивирующие вирусные, паразитарные, вирусные инфекции
Судороги
Дисморфия лица
Другие пороки развития –атрезия
Синдром
Ди Джорджи
Клиника:
Рецидивирующие вирусные, паразитарные, вирусные инфекции
Судороги
Дисморфия лица
Другие пороки развития –атрезия

Синдром Незелофа
Гипоплазия тимуса
Может наследоваться по аутосомно-рецессивному пути и быть
Синдром Незелофа
Гипоплазия тимуса
Может наследоваться по аутосомно-рецессивному пути и быть
Синдром Незелофа
Синдром Незелофа

Дефицит В-клеточного звена
Агамма(гипогамма)глобулинемия (синдром Брутона) рецессивный тип наследования, встречается в основном
Дефицит В-клеточного звена
Агамма(гипогамма)глобулинемия (синдром Брутона) рецессивный тип наследования, встречается в основном

Синдром Брутона
Снижено количество иммуноглобулинов всех классов, но в большей степени IgG
Отсутствуют
Синдром Брутона
Снижено количество иммуноглобулинов всех классов, но в большей степени IgG
Отсутствуют

клиника
Определяется на 5-9 месяце жизни
Рецидивирующие гнойные инфекции
Менингиты
Отсутствие реакции лф.узлов и селезенки
клиника
Определяется на 5-9 месяце жизни
Рецидивирующие гнойные инфекции
Менингиты
Отсутствие реакции лф.узлов и селезенки

Дефицит IgA
Различают недостаточность полную или селективную (IgA2)
Развиваются частые респираторные инфекции
Заместительная терапия
Дефицит IgA
Различают недостаточность полную или селективную (IgA2)
Развиваются частые респираторные инфекции
Заместительная терапия

Х-сцепленная агаммаглобулинемия с гиперглобулинемией М
Развитие связано с невозможностью переключения синтеза иммуноглобулинов
Х-сцепленная агаммаглобулинемия с гиперглобулинемией М
Развитие связано с невозможностью переключения синтеза иммуноглобулинов

Х-сцепленная агаммаглобулинемия с гиперглобулинемией М
Лабораторные данные:
IgA, IgG,IgE – не определяются
Уровень IgM
Х-сцепленная агаммаглобулинемия с гиперглобулинемией М
Лабораторные данные:
IgA, IgG,IgE – не определяются
Уровень IgM

ТКИД (швейцарский тип)
Х-сцепленный тип
Дефект на уровне стволовой клетки
нежизнеспособность
ТКИД (швейцарский тип)
Х-сцепленный тип
Дефект на уровне стволовой клетки
нежизнеспособность

Комбинированные ИД
Синдром Вискотта-Олдрича
Нарушение активации Т-h и Т-s
<концентрация IgM, (IgA IgE >)
Комбинированные ИД
Синдром Вискотта-Олдрича
Нарушение активации Т-h и Т-s
<концентрация IgM, (IgA IgE >)

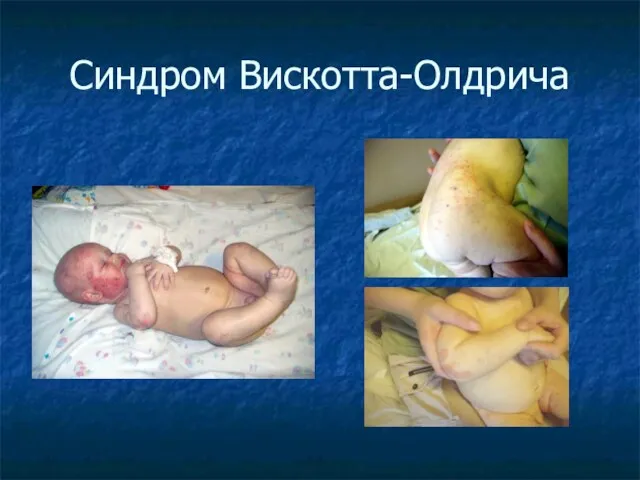
Синдром Вискотта-Олдрича
Клиника:
Экзема
Тромбоцитопения
Бактериальные и вирусные инфекции
Синдром Вискотта-Олдрича
Клиника:
Экзема
Тромбоцитопения
Бактериальные и вирусные инфекции
Синдром Вискотта-Олдрича
Синдром Вискотта-Олдрича

Синдром Луи-Бар
(атаксия-телеангиэктазия)
Гипоплазия тимуса
Гипоплазия лимф. узлов, селезенки
Количественная и функциональная недостаточность Т-лимфоцитов
Снижение количества
Синдром Луи-Бар
(атаксия-телеангиэктазия)
Гипоплазия тимуса
Гипоплазия лимф. узлов, селезенки
Количественная и функциональная недостаточность Т-лимфоцитов
Снижение количества

Синдром Луи-Бар
Клиника:
Атаксия
Телеангиэктазы
(на носу, ушах, конъюктиве)
Рецидивирующие инфекции
Опухолевые заболевания
Синдром Луи-Бар
Клиника:
Атаксия
Телеангиэктазы
(на носу, ушах, конъюктиве)
Рецидивирующие инфекции
Опухолевые заболевания
 Преждевременные роды. Определение. Причины. Патогенетические аспекты
Преждевременные роды. Определение. Причины. Патогенетические аспекты Острые отравления у детей
Острые отравления у детей Догляд за недоношеними дітьми після народження
Догляд за недоношеними дітьми після народження Физическое развитие
Физическое развитие Тема 9
Тема 9 Оценка комплексного состояния здоровья ребенка, определение группы здоровья по форме 112/у
Оценка комплексного состояния здоровья ребенка, определение группы здоровья по форме 112/у Организация лечебноэвакуационного обеспечения населения в чрезвычайных ситуациях
Организация лечебноэвакуационного обеспечения населения в чрезвычайных ситуациях Попередження вірусних захвоорювань
Попередження вірусних захвоорювань Средства физической культуры в регулировании работоспособности
Средства физической культуры в регулировании работоспособности Доброкачественные опухоли и гиперпластические процессы печени
Доброкачественные опухоли и гиперпластические процессы печени Нарушения водно-электролитного обмена
Нарушения водно-электролитного обмена Безпека харчування. Вибір харчових продуктів
Безпека харчування. Вибір харчових продуктів Диабетические комы
Диабетические комы Клещевой энцефалит и его профилактика
Клещевой энцефалит и его профилактика Вирустар генетикасы. Вирустық геномның ұйымдасуы. Вирустық геномдардың репликациясы
Вирустар генетикасы. Вирустық геномның ұйымдасуы. Вирустық геномдардың репликациясы Psychopharmacologie – troubles anxieux
Psychopharmacologie – troubles anxieux Ерік. Балада ерікті дамыту жолдары
Ерік. Балада ерікті дамыту жолдары Сердечно-легочная реанимация (СЛР)
Сердечно-легочная реанимация (СЛР) Амебиаз (amoebiasis). Этиология. Эпидемилогия
Амебиаз (amoebiasis). Этиология. Эпидемилогия Лучевая диагностика костно-суставной системы
Лучевая диагностика костно-суставной системы Синдром портальной гипертензии
Синдром портальной гипертензии Патогенна дія хімічних та біологічних факторів на організм. Роль спадковості в патології. (Лекція 4)
Патогенна дія хімічних та біологічних факторів на організм. Роль спадковості в патології. (Лекція 4) Trypanosomiasis
Trypanosomiasis Радиоизотопная диагностика
Радиоизотопная диагностика Оказание медицинской помощи при переломах
Оказание медицинской помощи при переломах Психотропные лекарственные средства угнетающего типа: нейролептики, транквилизаторы, седативные
Психотропные лекарственные средства угнетающего типа: нейролептики, транквилизаторы, седативные Теміртапшылықты анемия кезіндегі зертханалық тексерулер
Теміртапшылықты анемия кезіндегі зертханалық тексерулер Бабезіоз великої рогатої худоби
Бабезіоз великої рогатої худоби