- Моногенные заболевания

Содержание
- 2. Моногенные болезни подчиняются менделевскому наследованию, в их основе лежат единичные генные или точковые мутации по типу
- 3. Моногенные болезни по преимущественному поражению вида обмена: болезни аминокислотного обмена; болезни углеводного обмена; болезни липидного; болезни
- 4. В результате мутации гена на молекулярном уровне возможны следующие варианты: 1) синтез аномального белка; 2) выработка
- 5. Снижение активности фермента А В С А1,А2 фермент 1 фермент 2 ген1 Снижение количества продуктов реакции
- 6. Клинические проявления генных болезней, тяжесть и скорость их развития зависят от особенностей генотипа организма (гены-модификаторы, доза
- 7. Особенностью генных (как и вообще всех наследственных) болезней является их гетерогенность. Это означает, что одно и
- 8. Диагностика наследственных болезней обмена веществ Ген Белок Метаболиты ДНК-диагностика Энзимодиагностика и другие методы анализа белков Хроматографические
- 9. Aутосомно-доминантный тип наследования
- 10. При аутосомно-доминантном типе наследования гетерозиготное носительство мутации оказывается достаточным для проявления заболевания. При этом мальчики и
- 11. Частота некоторых аутосомно-доминантный заболеваний и % новых случаев
- 12. 2.2 Аутосомно-рецессивный тип наследования
- 13. 2.2 Аутосомно-рецессивный тип наследования
- 14. Аутосомно-рецессивный тип наследования с кровнородственными браками
- 15. Аутосомно-рецессивный тип наследования с кровнородственными браками
- 16. Аутосомно-рецессивные при браке двух гетерозиготных носителей одного и того же мутантного рецессивного гена в среднем 50%
- 17. Частота некоторых рецессивных заболеваний и частота гетерозиготного носительства
- 18. 2.3 X-сцепленный
- 20. Родословная Царской семьи
- 22. X-сцепленный рецессивный
- 23. Х-сцепленный рецессивный заболевание наблюдается у мужчин-родственников пробанда по материнской линии; сыновья никогда не наследуют заболевание отца;
- 24. X-сцепленный рецессивный несахарный диабет дефицит глюкозо-6-фосфат-дегидрогеназы мышечная дистрофия Дюшена гемофилия А, В ихтиоз синдром Аарскога
- 25. 2.4 X-сцепленный доминантный
- 26. 2.4 X-сцепленный доминантный
- 27. Х-сцепленный доминантный у больного пробанда обязательно болен один из родителей; у больного отца все дочери больны,
- 28. Х-сцепленный доминантный фосфатдиабет синдром Ретта синдром Коффина-Лоури синдромГольца и др.
- 29. 2.5 Y-сцепленный тип
- 30. Y-сцепленное наследование в Y-хромосоме находятся гены: детерминирующий развитие семенников, отвечающий за сперматогенез (фактор азооспермии), контролирующий интенсивность
- 31. 2.6 Митохондриальная наследственность
- 32. 2.6 Митохондриальная наследственность
- 33. Цитоплазматическая наследственность атрофия зрительного нерва Лебера; митохондриальная миоэнцефалопатия; синдром Лея; болезнь Кернса—Сейра Т.к. изменения митохондриального генома
- 34. Наследственные болезни аминокислотного обмена
- 35. Фенилкетонурия
- 36. У больных нарушено превращение аминокислоты фенилаланина в тирозин из-за резкого снижения активности фермента фенилаланингидроксилазы. В результате
- 37. Фенилкетонурия встречается в среднем в мировом масштабе с частотой 1 на 1000 новорожденных. Локус (фенилгидроксилазы) расположен
- 38. Ребенок с фенилкетонурией рождается здоровым, но в первые же недели в связи с поступлением фенилаланина в
- 40. Большинство больных - блондины со светлой кожей и голубыми глазами, что определяется недостаточным синтезом пигмента меланина
- 41. Диагностика Производится полуколичественным тестом или количественным определением фенилаланина в крови. При нелеченных случаях возможно выявление продуктов
- 42. Лечение и профилактика При своевременной диагностике патологических изменений можно полностью избежать, если с рождения и до
- 43. Лечение и профилактика При рождении ребёнка в роддомах на 3-4 сутки берут анализ крови и проводят
- 44. Лечение и профилактика Лечение проводится в виде строгой диеты от обнаружения заболевания как минимум до полового
- 46. Некоторые (мягкие) формы заболевания поддаются лечению кофактором (тетрагидробиоптерином) пораженного фермента (фенилаланингидроксилазы). Разрабатываются новые подходы к лечению
- 47. Наследственные болезни соединительной ткани
- 48. Синдром Марфана
- 49. Синдром Марфана (Болезнь Марфана, Marfan syndrome) — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром
- 50. Причина болезни -- мутация в гене, ответственном за синтез белка соединительнотканных волокон фибриллина. Блокирование его синтеза
- 52. В классических случаях лица с синдромом Марфана высоки(долихостено-мелия), имеют удлиненные конечности, вытянутые пальцы (арахнодактилия) и недоразвитие
- 53. Арахнодактилия
- 54. Помимо характерных изменений в органах опорно-двигательного аппарата, наблюдается патология в органах зрения и сердечно-сосудистой системы, что
- 55. Тест запястья
- 56. Тест большого пальца
- 57. Больных с синдромом Марфана отличают высокий рост, длинные паукообразные пальцы, деформация грудной клетки (воронкообразная, килевидная, уплощенная),
- 58. Нередко имеют место ухудшение зрения, изменение формы и размера хрусталика, значительная миопия вплоть до отслойки сетчатки,
- 59. Лечение в основном симптоматическое. Положительное действие оказывают массаж, лечебная физкультура, а в ряде случаев оперативное вмешательство.
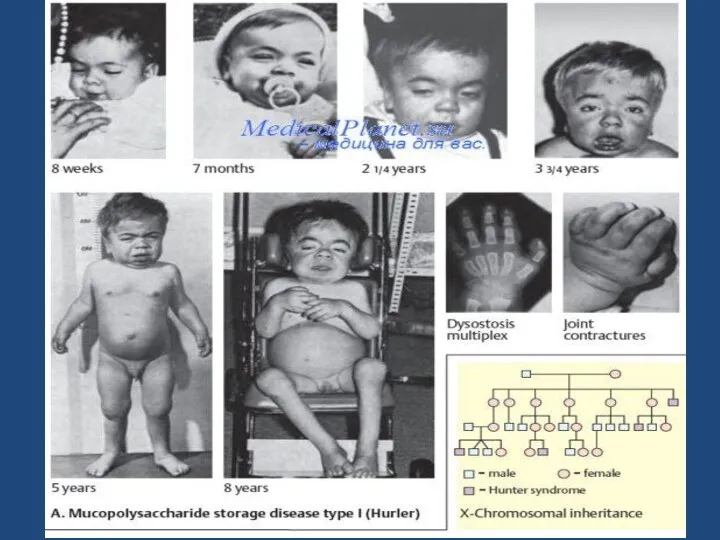
- 61. МУКОПОЛИСАХАРИДОЗ
- 63. Мукополисахаридозы Группа наследственных (генетических) болезней, вызванных аномалиями обмена мукополисахаридов и проявляющихся различными дефектами костной, хрящевой, соединительной
- 64. ЭТИОЛОГИЯ генетический дефект ферментного расщепления углеводной части молекулы мукополисахаридов (гликозоаминогликанов), в тканях (преимущественно в фибробластах и
- 65. Симптомокомплексы Нарушается функциональное состояние различных органов и систем, а поскольку гликозаминогликаны входят в состав соединительной ткани,
- 66. ТИПЫ 1 ТИП- СИНДРОМ ГУРЛЕР 2 ТИП- СИНДРОМ ХАНТЕРА 3 ТИП- СИНДРОМ САНФИЛИППО 4 ТИП- СИНДРОМ
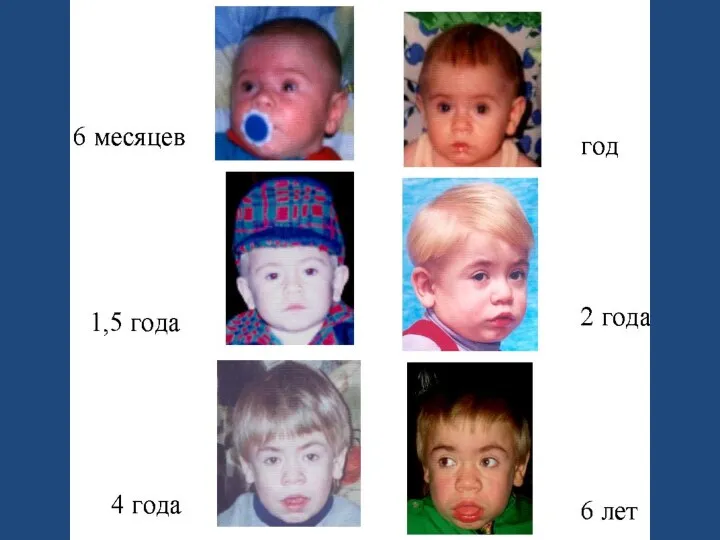
- 67. 1 ТИП- СИНДРОМ ГУРЛЕР Голова увеличена Выражены лобные бугры Шея почти отсутствует Рост резко уменьшен Язык
- 68. Грудная клетка укорочена Ограничена подвижность в суставах, контрактуры Гепатоспленомегалия Пупочная и паховая грыжи Склонность к хроническим
- 70. НЕВРОЛОГИЧЕСКИЙ СТАТУС гипертензионно-гидроцефальный синдром диффузная мышечная гипотония повышение сухожильных рефлексов общая двигательная заторможенность снижение интеллекта и
- 71. ОФТАЛЬМОЛОГИЧЕСКИЕ СИМПТОМЫ Гипертелоризм Густые ресницы Пастозные веки Макрокорнеа Конъюнктива век и глазного яблока цианотична, отечна Утолщение
- 72. СЕРДЕЧНО- СОСУДИСТАЯ СИСТЕМА Характерны изменения со стороны - клапанов сердца - миокарда - эндокарда - крупных
- 73. МПС 2 ТИПА - СИНДРОМ ХАНТЕРА ТИП НАСЛЕДОВАНИЯ – Х-сцепленный ЧАСТОТА ВСТРЕЧАЕМОСТИ - 1:70000
- 74. МПС 2 ТИПА - СИНДРОМ ХАНТЕРА недостаточность сульфо-идуронат сульфатазы • Подтип 2А – тяжёлое течение •Подтип
- 76. ПРОГРАММА ОБСЛЕДОВАНИЯ БОЛЬНЫХ С МПС Генеалогический метод Метод клинического анализа Рентгено-функциональные методы Биохимические методы Морфологические методы
- 77. КЛИНИЧЕСКАЯ ДИАГНОСТИКА Выделение главных симптомов и признаков, присущих каждому типу МПС
- 78. ЛАБОРАТОРНАЯ ДИАГНОСТИКА Скрининг- тесты (суточная моча на ГАГ – проводится в лаборатории Медико-генетической консультации НОДКБ) Определение
- 80. ЛЕЧЕНИЕ 1. СИМПТОМАТИЧЕСКАЯ ТЕРАПИЯ -МЕДИКАМЕНТОЗНАЯ -ХИРУРГИЧЕСКАЯ -ФИЗИОТЕРАПЕВТИЧЕСКАЯ 2. ЗАМЕСТИТЕЛЬНАЯ ПАТОГЕНЕТИЧЕСКАЯ ТЕРАПИЯ -АЛЬДУРАЗИМ -ЭЛАПРАЗА -НАГЛАЗИМ 3. ТРАНСПЛАНТАЦИЯ
- 81. ИСХОДЫ Имеются данные, о том что при легких формах МПС больные могут дожить до 50-60 лет,
- 82. ПРОФИЛАКТИКА МПС Медико-генетическое консультирование семей Выявление гетерозиготных носителей Пренатальная диагностика- определение активности лизосомных ферментов в биоптатах
- 83. Наследственные нарушения обмена в эритроцитах К этой группе относятся болезни, связанные чаще всего с укорочением срока
- 84. Гемоглобин -- основной белок эритроцитов. В настоящее время хорошо изучена аминокислотная последовательность и структура его молекулы.
- 85. В результате мутаций в эритроцитах и гемоглобине возникают наследственные болезни человека : гемолитические анемии и гемоглобинопатии.
- 86. Гемолитические анемии включают заболевания, обусловленные снижением уровня гемоглобина и укорочением срока жизни эритроцитов. Кроме того, причиной
- 87. Серповидноклеточная анемия — это наследственная гемоглобинопатия, связанная с таким нарушением строения белка гемоглобина, при котором он
- 88. Эритроциты, несущие гемоглобин S вместо нормального гемоглобина А, под микроскопом имеют характерную серпообразную форму (форму серпа),
- 89. Эритроциты, несущие гемоглобин S, обладают пониженной стойкостью и пониженной кислород-транспортирующей способностью, поэтому у больных с серповидноклеточной
- 90. Серповидноклеточная анемия наследуется по аутосомно-рецессивному типу (с неполным доминированием)
- 91. У носителей, гетерозиготных по гену серповидноклеточной анемии, в эритроцитах присутствуют примерно в равных количествах гемоглобин S
- 92. При этом в нормальных условиях у носителей симптомы практически никогда не возникают, и серповидные эритроциты выявляются
- 93. Симптомы у носителей могут появиться при гипоксии (например, при подъеме в горы) или тяжелой дегидратации организма.
- 94. Серповидноклеточная анемия весьма распространена в регионах мира, эндемичных по малярии, причем больные серповидноклеточной анемией обладают повышенной
- 95. Повышенной устойчивостью к малярии обладают и гетерозиготы-носители, которые анемией не болеют (преимущество гетерозигот), что объясняет высокую
- 96. Симптомы Усталость и анемия Приступы боли Отек и воспаление пальцев рук и/или ног и артрит Бактериальные
- 97. Симптомы серповидноклеточной анемии делятся на две основные категории. Из-за хрупкости красных клеток крови всегда наблюдается анемия,
- 98. Кроме этого, периодическая закупорка мелких капилляров в любой части тела может привести к широкому спектру различных
- 99. Обычно никаких симптомов не проявляется до 3-месячного возраста. Первыми признаками серповидноклеточной анемии у младенца обычно являются
- 100. Единственным очень серьёзным осложнением серповидноклеточной анемии у ребенка до 5-летнего возраста является инфекция. Скопление эритроцитов и
- 101. Проблемой детей школьного возраста с серповидноклеточной анемией обычно является эпизодическая закупорка эритроцитами капилляров больших костей. В
- 102. С возрастом процесс закупорки капилляров может затрагивать и другие органы. Если это произойдет, например, в легких,
- 103. У взрослых с серповидноклеточной анемией могут обнаруживаться симптомы хронической (постоянной или длительной) закупорки капилляров легких и
- 104. Специальных методов лечения нет. Важное значение имеет предохранение больного от воздействия факторов, провоцирующих развитие болезни (гипоксия,
- 105. Болезнь Вильсона — Коновалова (гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона — Коновалова)
- 106. Болезнь Вильсона — Коновалова врождённое нарушение метаболизма меди, приводящее к тяжелейшим наследственным болезням центральной нервной системы
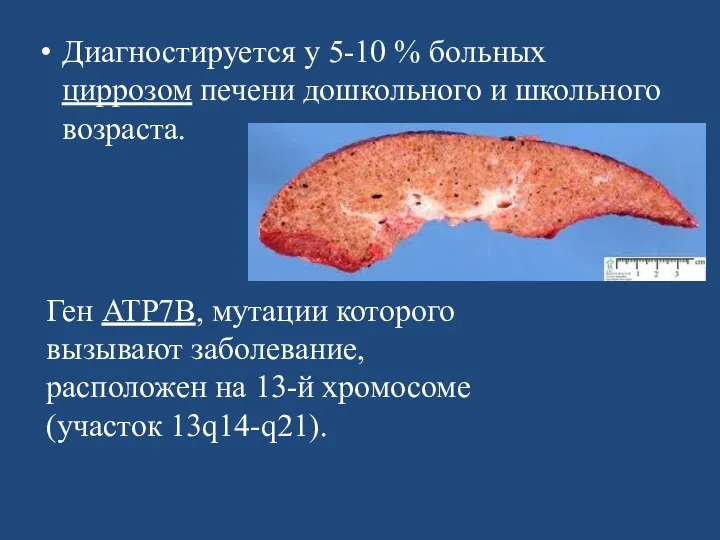
- 107. Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Ген ATP7B, мутации которого вызывают
- 108. Заболевание передается по аутосомно-рецессивному типу.
- 109. Основную роль в патогенезе играет нарушение обмена меди, её накопление в нервной (особенно поражены базальные ганглии),
- 110. В печени формируется крупноузловой или смешанный цирроз. В почках в первую очередь страдают проксимальные канальцы.
- 111. В головном мозге поражаются в большей степени базальные ганглии, зубчатое вещество и черная субстанция.
- 112. Отложение меди в десцементовой мембране глаза приводит к формированию кольца Кайзера-Флейшера.
- 113. Гепато-церебральная дистрофия начинается в детском или молодом возрасте и имеет хроническое прогрессирующее течение. Во многих случаях
- 114. Со стороны нервной системы на первый план выступают экстрапирамидные симптомы в виде мышечной ригидности, гиперкинезов и
- 115. Типичным симптомом болезни является кольцо Кайзера-Флейшера — отложение по периферии роговой оболочки содержащего медь зеленовато-бурого пигмента;
- 116. Иногда отмечается желтовато-коричневая пигментация кожи туловища и лица. Часты геморрагические явления (кровоточивость дёсен, носовые кровотечения, положительная
- 117. Лечение Патогенетическое лечение при гепатолентикулярной дегенерации направлено на увеличение выведения меди из организма. Для этого применяются
- 118. ГЕМОФИЛИЯ
- 119. Гемофили́я — наследственное заболевание, связанное с нарушением коагуляции ; при этом заболевании возникают кровоизлияния в суставы,
- 120. При гемофилии резко возрастает опасность гибели пациента от кровоизлияния в мозг и другие жизненно важные органы,
- 121. Гемофилия появляется из-за изменения одного гена в хромосоме X. Различают три типа гемофилии (A, B, C).
- 122. Гемофилия A (рецессивная мутация в X-хромосоме) вызывает недостаточность в крови необходимого белка — так называемого фактора
- 123. ГЕМОФИЛИЯ A
- 124. Обычно болезнью страдают мужчины (наследование, сцепленное с полом), женщины же обычно выступают как носительницы гемофилии и
- 125. Общеизвестным является мнение, что женщины не болеют гемофилией, однако это мнение ошибочно. Такое событие крайне маловероятно,
- 126. Кроме того, примерно в 15-25 % случаев обследование матерей мальчиков, страдающих гемофилией, не выявляет указанных мутаций
- 127. Самой известной носительницей гемофилии в истории была королева Виктория; по-видимому, эта мутация произошла в её генотипе
- 128. Гемофилией страдал один из сыновей Виктории (Леопольд, герцог Олбани), а также ряд внуков и правнуков (родившихся
- 129. Ведущими симптомами гемофилии А и В являются повышенная кровоточивость с первых месяцев жизни; подкожные, межмышечные, субфасциальные,
- 130. обильные посттравматические кровотечения; гемартрозы крупных суставов, с вторичными воспалительными изменениями, которые приводят к формированию контрактур и
- 132. Наиболее распространенное заблуждение о гемофилии — это то, что больной гемофилии может истечь кровью от малейшей
- 133. Для диагностики гемофилии применяется: коагулограмма, определение времени свёртываемости, добавление образцов плазмы с отсутствием одного из факторов
- 134. ЛЕЧЕНИЕ Хотя болезнь на сегодняшний день неизлечима, её течение контролируется с помощью инъекций недостающего фактора свёртываемости
- 135. На настоящий момент для лечения используются концентраты факторов свертывания как полученные из донорской крови, так и
- 136. Гемофилия B (рецессивная мутация в X-хромосоме) недостаточность фактора крови IX (Кристмаса). Нарушено образование вторичной коагуляционной пробки.
- 137. Муковисцидóз (кистозный фиброз)
- 138. Муковисцидóз (кистозный фиброз) — системное наследственное заболевание, обусловленное мутацией гена трансмембранного регулятора муковисцидоза и характеризующееся поражением
- 139. Патологический ген локализуется в середине длинного плеча 7-й хромосомы. Муковисцидоз наследуется по аутосомно-рецессивному типу и регистрируется
- 140. Различают следующие клинические формы муковисцидоза: преимущественно лёгочная форма (респираторная, бронхолёгочная); преимущественно кишечная форма; смешанная форма с
- 141. Патологические изменения в лёгких характеризуются признаками хронического бронхита с развитием бронхоэктазов и диффузного пневмосклероза.
- 142. В просвете бронхов находится вязкое содержимое слизисто-гнойного характера. Нередкой находкой являются ателектазы и участки эмфиземы.
- 143. У многих больных течение патологического процесса в лёгких осложняется наслоением бактериальной инфекции (патогенный золотистый стафилококк, гемофильная
- 144. В поджелудочной железе выявляется диффузный фиброз, утолщение междольковых соединительноткан-ных прослоек, кистозные изменения мелких и средних протоков.
- 145. В печени отмечается очаговая или диффузная жировая и белковая дистрофия клеток печени, желчные стазы в междольковых
- 146. При мекониевой непроходимости выражена атрофия слизистого слоя, просвет слизистых желез кишечника расширен, заполнен эозинофильными массами секрета,
- 147. Нередко муковисцидоз сочетается с различными пороками развития желудочно-кишечного тракта
- 148. Диагностика муковисцидоза ДНК-диагностика наиболее чувствительная и специфическая. Ложные результаты получают в 0,5—3 % случаев.
- 149. Лечение муковисцидоза симптоматическое. коррекция нарушенной функции поджелудочной железы путём применения панкреатина или комбинированных препаратов, содержащих наряду
- 150. Критерием качества диагностики и лечения муковисцидоза является средняя продолжительность жизни больных. В европейских странах этот показатель
- 151. Нейрофиброматоз
- 152. Нейрофиброматоз — заболевание из группы факоматозов. Существует 7 типов нейрофиброматоза. Нейрофиброматоз I типа (НФ1) Основными симптомами
- 153. Нейрофиброматоз II типа (НФ2) Основными симптомами НФ2 являются: двусторонняя невринома VIII нерва; наличие родственника, поражённого НФ2
- 154. III тип — редкая форма нейрофиброматоза, характеризуется ладонными нейрофибромами, бледноватыми относительно большими пятнами цвета кофе с
- 155. Нейрофиброматоз I (первого) типа ( болезнь фон Реклингхаузена, синдром Реклингхаузена, NF-1) самое распространённое наследственное заболевание, предрасполагающее
- 156. Основными симптомами НФ1 являются: наличие множества светло-коричневых пятен на коже (от 5 до 15 мм); наличие
- 157. Тип наследования
- 158. Клиническая картина наличие пигментных пятен на коже цвета «кофе с молоком», нейрофибром, большинство из которых располагаются
- 159. Нейрофибромы чаще локализуются по ходу периферических нервов. Однако может поражаться спинной и головной мозг, находят нейрофибромы
- 160. Нейрофибромы Для данного заболевания характерно появление большого количества нейрофибром, как кожных, так и плексиформных. Кожные нейрофибромы
- 161. Плексиформные нейрофибромы развиваются на крупных нервах и приводят к нарушению их функций. Также плексиформные нейрофибромы характеризуются
- 162. Опухоли центральной нервной системы Наиболее часто возникающими при данном заболевании опухолями ЦНС являются глиомы зрительных нервов,
- 163. Пигментные нарушения пигментные пятна носят характер пятен цвета «кофе с молоком» (фр. cafe-au-lait, англ. milk coffee)
- 164. Узелки Лиша Узелки Лиша встречаются практически у всех больных нейрофиброматозом I типа старше 20 лет. Они
- 165. Костные изменения
- 166. МРТ левой голени: злокачественная опухоль оболочки большеберцового нерва при синдроме Реклингхаузена (NF-1).
- 167. Лечение Лечение оперативное. Показаниями для него являются резкая болезненность или изъязвление опухоли, затруднение движений, сдавление или
- 168. Нейрофиброматоз II типа Возникающие при нейрофиброматозе II типа опухоли — доброкачественные, но более биологически агрессивные по
- 169. Основными симптомами НФ2 являются: двусторонняя невринома VIII нерва;
- 170. Интрамедуллярные и экстрамедуллярные спиальные опухоли у пациентов с НФ II
- 171. Лечение Хирургическое. При двусторонних невриномах и сохранном слухе, лечение рекомендуется начинать с опухоли меньшего размера, при
- 173. Скачать презентацию

Моногенные болезни
подчиняются менделевскому наследованию, в их основе лежат единичные генные или
Моногенные болезни
подчиняются менделевскому наследованию, в их основе лежат единичные генные или

Моногенные болезни
по преимущественному поражению вида обмена:
болезни аминокислотного обмена;
болезни углеводного
Моногенные болезни
по преимущественному поражению вида обмена:
болезни аминокислотного обмена;
болезни углеводного

В результате мутации гена на молекулярном уровне возможны следующие варианты:
1)
В результате мутации гена на молекулярном уровне возможны следующие варианты:
1)
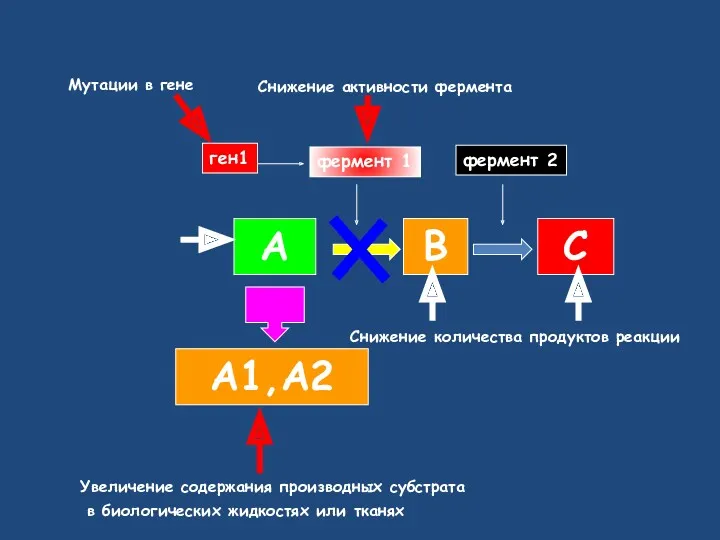
Снижение активности фермента
А
В
С
А1,А2
фермент 1
фермент 2
ген1
Снижение количества продуктов реакции
Мутации в
Снижение активности фермента
А
В
С
А1,А2
фермент 1
фермент 2
ген1
Снижение количества продуктов реакции
Мутации в

Клинические проявления генных болезней, тяжесть и скорость их развития зависят от
Клинические проявления генных болезней, тяжесть и скорость их развития зависят от

Особенностью генных (как и вообще всех наследственных) болезней является их гетерогенность.
Особенностью генных (как и вообще всех наследственных) болезней является их гетерогенность.
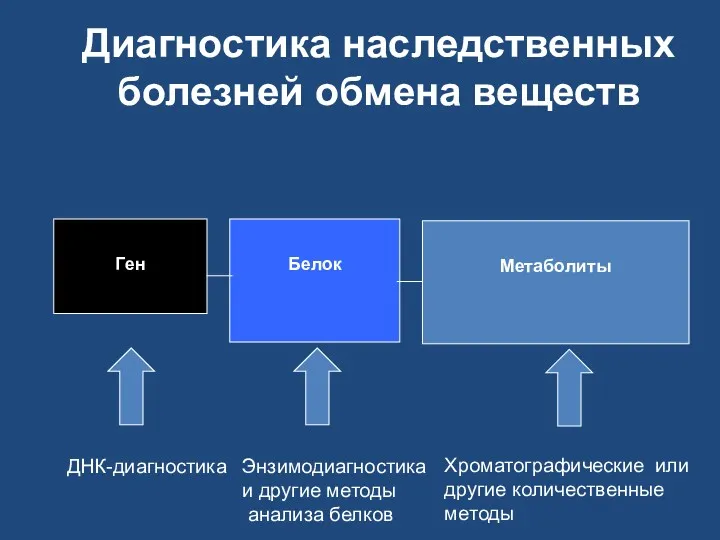
Диагностика наследственных болезней обмена веществ
Ген
Белок
Метаболиты
ДНК-диагностика
Энзимодиагностика
и другие методы
анализа белков
Хроматографические
Диагностика наследственных болезней обмена веществ
Ген
Белок
Метаболиты
ДНК-диагностика
Энзимодиагностика
и другие методы
анализа белков
Хроматографические
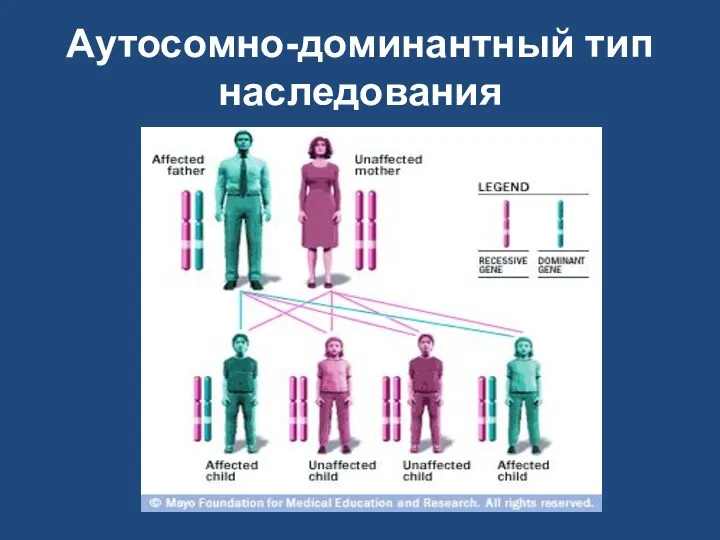
Aутосомно-доминантный тип наследования
Aутосомно-доминантный тип наследования

При аутосомно-доминантном типе наследования гетерозиготное носительство мутации оказывается достаточным для проявления
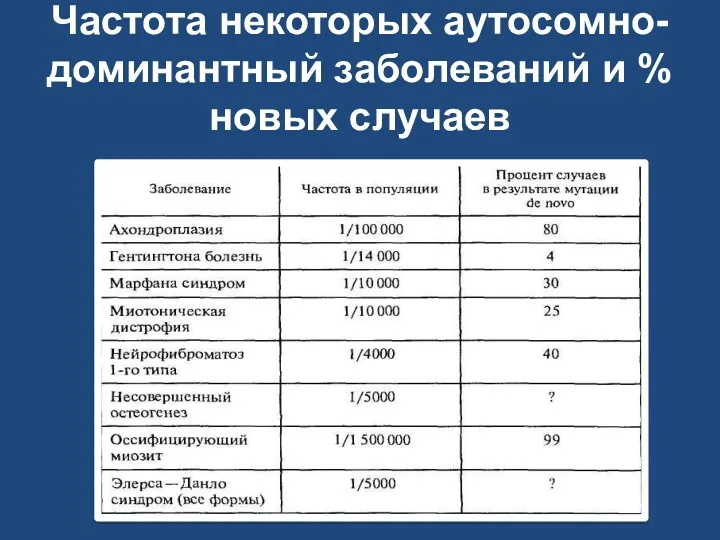
Частота некоторых аутосомно-доминантный заболеваний и % новых случаев
Частота некоторых аутосомно-доминантный заболеваний и % новых случаев
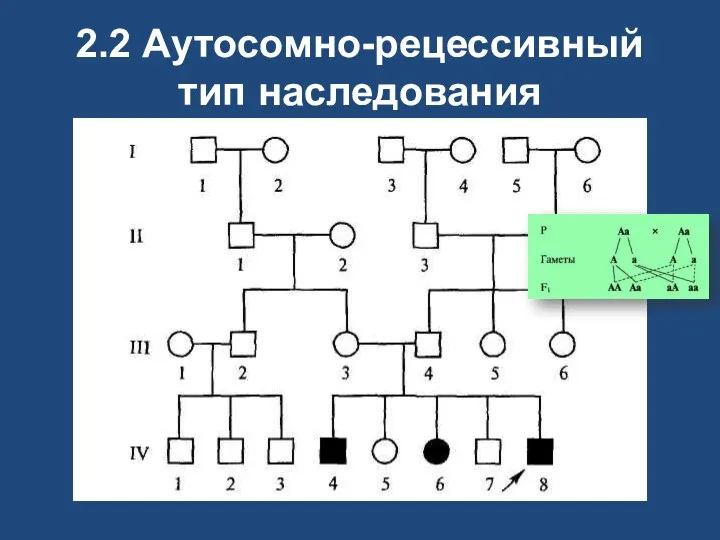
2.2 Аутосомно-рецессивный тип наследования
2.2 Аутосомно-рецессивный тип наследования
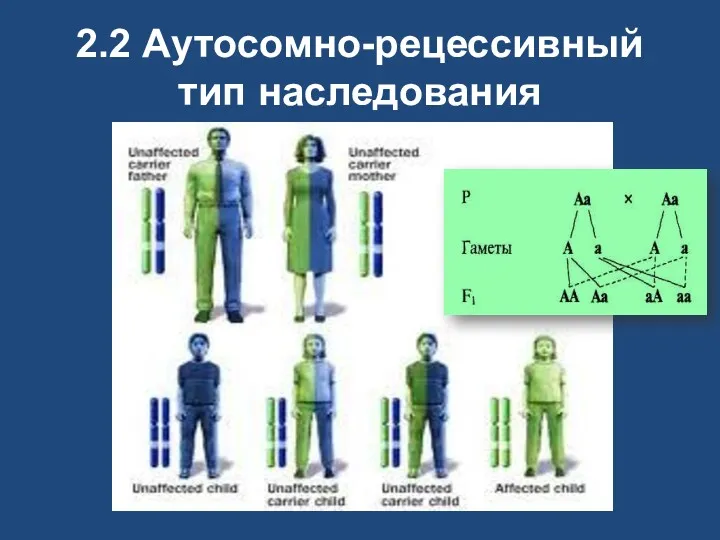
2.2 Аутосомно-рецессивный тип наследования
2.2 Аутосомно-рецессивный тип наследования
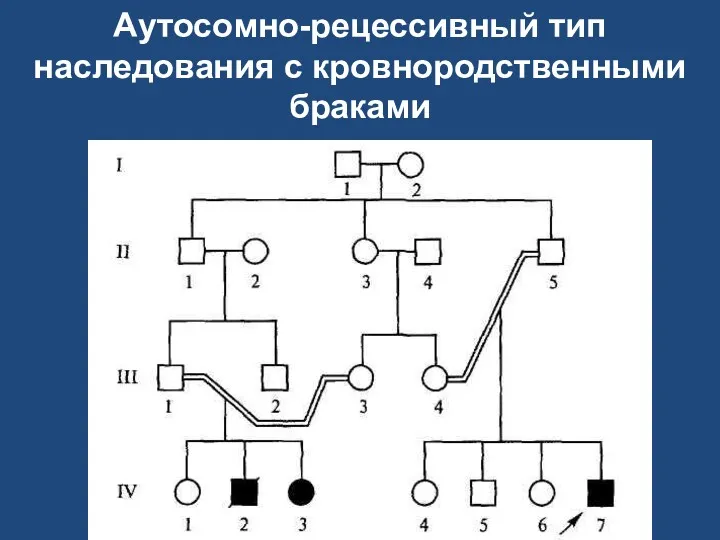
Аутосомно-рецессивный тип наследования с кровнородственными браками
Аутосомно-рецессивный тип наследования с кровнородственными браками
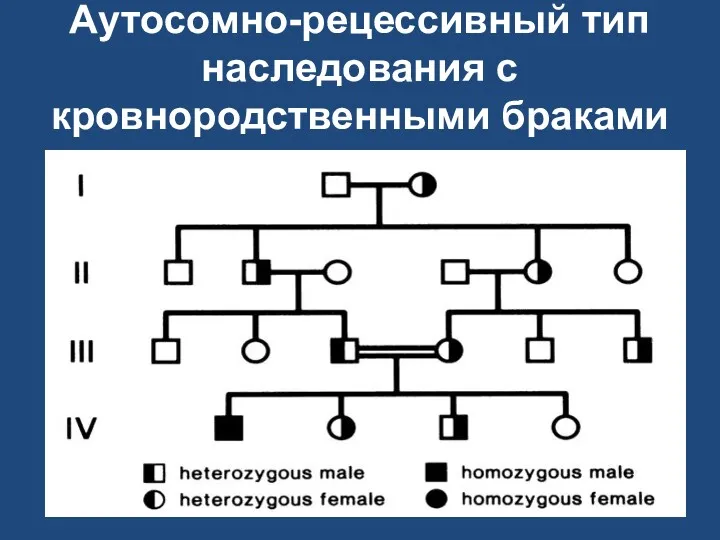
Аутосомно-рецессивный тип наследования с кровнородственными браками
Аутосомно-рецессивный тип наследования с кровнородственными браками

Аутосомно-рецессивные
при браке двух гетерозиготных носителей одного и того же мутантного рецессивного
Аутосомно-рецессивные
при браке двух гетерозиготных носителей одного и того же мутантного рецессивного

Частота некоторых рецессивных заболеваний и частота гетерозиготного носительства
Частота некоторых рецессивных заболеваний и частота гетерозиготного носительства
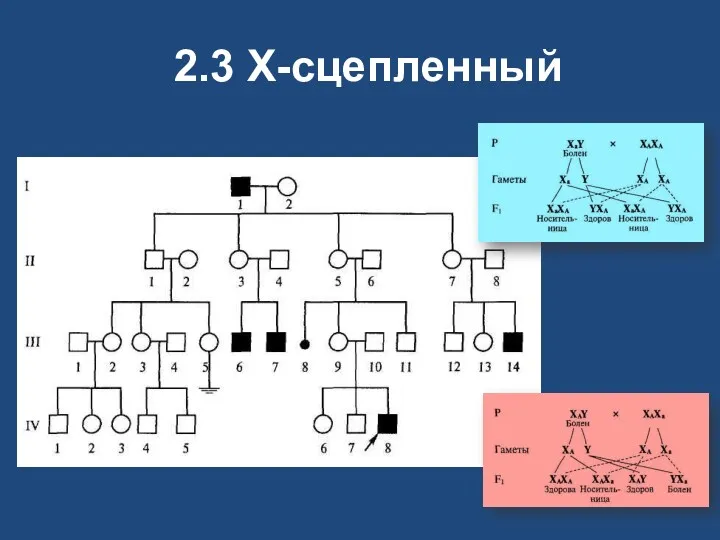
2.3 X-сцепленный
2.3 X-сцепленный

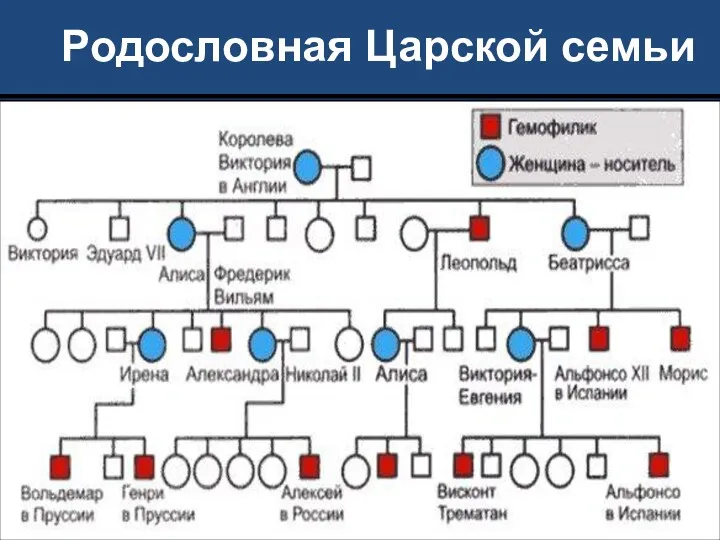
Родословная Царской семьи
Родословная Царской семьи

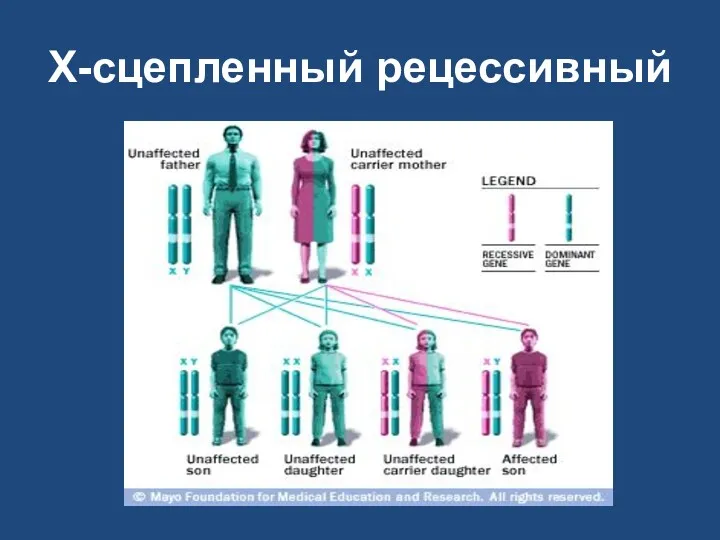
X-сцепленный рецессивный
X-сцепленный рецессивный

Х-сцепленный рецессивный
заболевание наблюдается у мужчин-родственников пробанда по материнской линии;
сыновья никогда не
Х-сцепленный рецессивный
заболевание наблюдается у мужчин-родственников пробанда по материнской линии;
сыновья никогда не

X-сцепленный рецессивный
несахарный диабет
дефицит глюкозо-6-фосфат-дегидрогеназы
мышечная дистрофия Дюшена
гемофилия А, В
ихтиоз
синдром Аарскога
X-сцепленный рецессивный
несахарный диабет
дефицит глюкозо-6-фосфат-дегидрогеназы
мышечная дистрофия Дюшена
гемофилия А, В
ихтиоз
синдром Аарскога
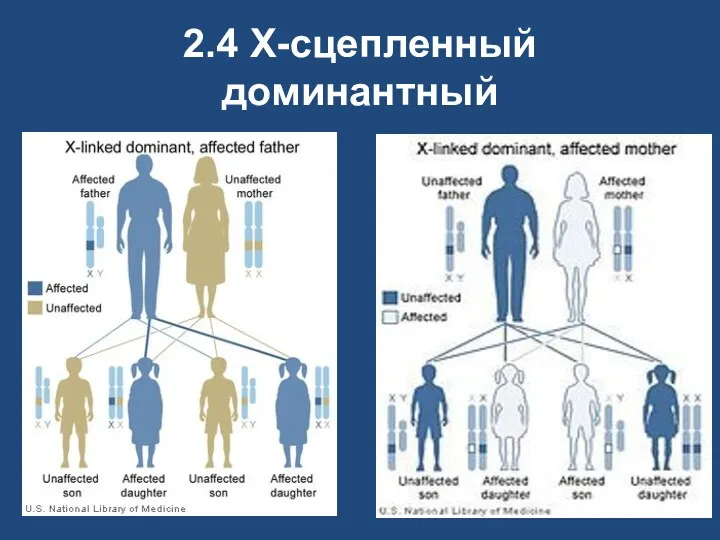
2.4 X-сцепленный доминантный
2.4 X-сцепленный доминантный
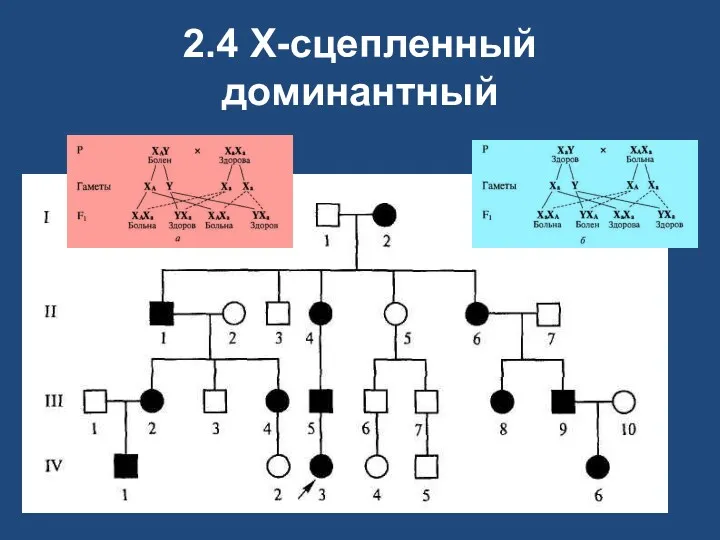
2.4 X-сцепленный доминантный
2.4 X-сцепленный доминантный

Х-сцепленный доминантный
у больного пробанда обязательно болен один из родителей;
у больного отца
Х-сцепленный доминантный
у больного пробанда обязательно болен один из родителей;
у больного отца

Х-сцепленный доминантный
фосфатдиабет
синдром Ретта
синдром Коффина-Лоури
синдромГольца
и др.
Х-сцепленный доминантный
фосфатдиабет
синдром Ретта
синдром Коффина-Лоури
синдромГольца
и др.
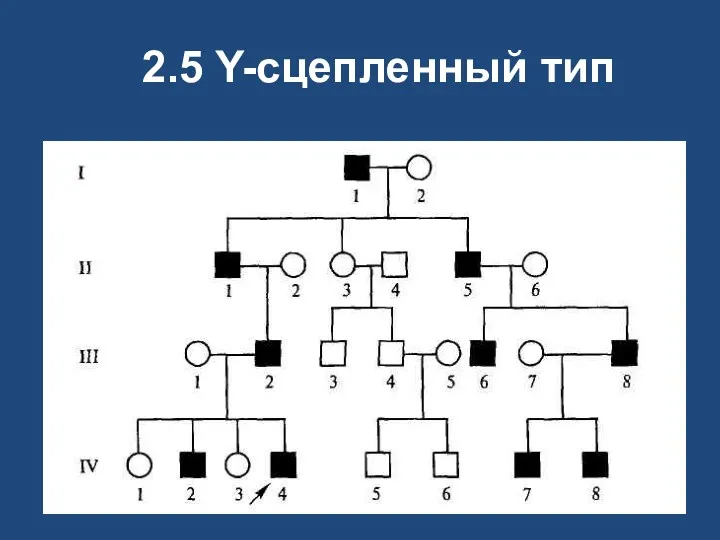
2.5 Y-сцепленный тип
2.5 Y-сцепленный тип

Y-сцепленное наследование
в Y-хромосоме находятся гены: детерминирующий развитие семенников, отвечающий за сперматогенез
Y-сцепленное наследование
в Y-хромосоме находятся гены: детерминирующий развитие семенников, отвечающий за сперматогенез

2.6 Митохондриальная наследственность
2.6 Митохондриальная наследственность
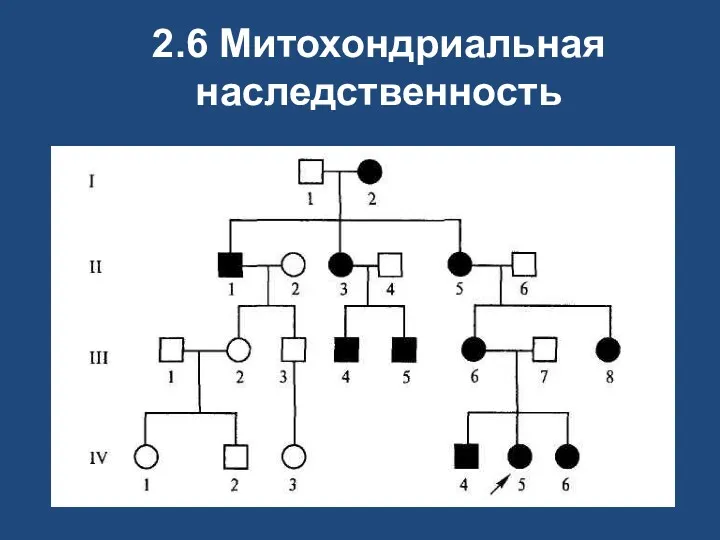
2.6 Митохондриальная наследственность
2.6 Митохондриальная наследственность

Цитоплазматическая наследственность
атрофия зрительного нерва Лебера;
митохондриальная миоэнцефалопатия;
синдром Лея;
болезнь Кернса—Сейра
Т.к. изменения митохондриального
Цитоплазматическая наследственность
атрофия зрительного нерва Лебера;
митохондриальная миоэнцефалопатия;
синдром Лея;
болезнь Кернса—Сейра
Т.к. изменения митохондриального

Наследственные болезни аминокислотного обмена
Наследственные болезни аминокислотного обмена

Фенилкетонурия
Фенилкетонурия

У больных нарушено превращение аминокислоты фенилаланина в тирозин из-за резкого снижения
У больных нарушено превращение аминокислоты фенилаланина в тирозин из-за резкого снижения

Фенилкетонурия встречается в среднем в мировом масштабе с частотой 1 на
Фенилкетонурия встречается в среднем в мировом масштабе с частотой 1 на

Ребенок с фенилкетонурией рождается здоровым, но в первые же недели в
Ребенок с фенилкетонурией рождается здоровым, но в первые же недели в
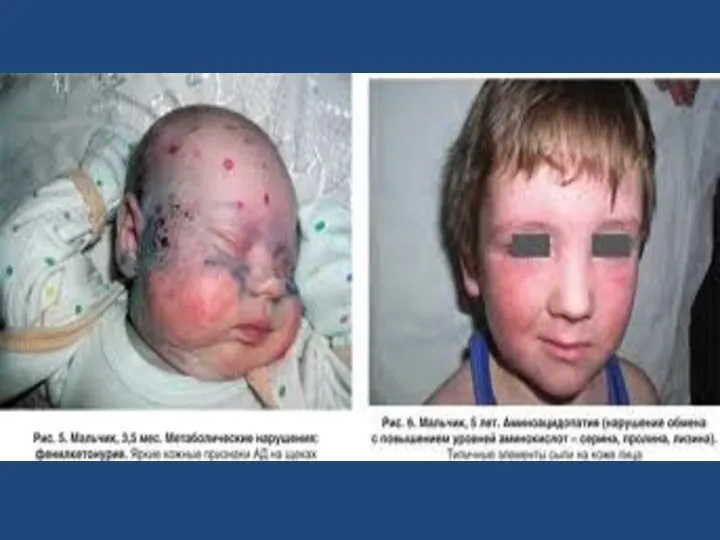

Большинство больных - блондины со светлой кожей и голубыми глазами, что
Большинство больных - блондины со светлой кожей и голубыми глазами, что

Диагностика
Производится полуколичественным тестом или количественным определением фенилаланина в крови.
При нелеченных
Диагностика
Производится полуколичественным тестом или количественным определением фенилаланина в крови.
При нелеченных

Лечение и профилактика
При своевременной диагностике патологических изменений можно полностью избежать, если
Лечение и профилактика
При своевременной диагностике патологических изменений можно полностью избежать, если

Лечение и профилактика
При рождении ребёнка в роддомах на 3-4 сутки берут
Лечение и профилактика
При рождении ребёнка в роддомах на 3-4 сутки берут

Лечение и профилактика
Лечение проводится в виде строгой диеты от обнаружения заболевания
Лечение и профилактика
Лечение проводится в виде строгой диеты от обнаружения заболевания


Некоторые (мягкие) формы заболевания поддаются лечению кофактором (тетрагидробиоптерином) пораженного фермента (фенилаланингидроксилазы).
Некоторые (мягкие) формы заболевания поддаются лечению кофактором (тетрагидробиоптерином) пораженного фермента (фенилаланингидроксилазы).

Наследственные болезни соединительной ткани
Наследственные болезни соединительной ткани

Синдром Марфана
Синдром Марфана

Синдром Марфана (Болезнь Марфана, Marfan syndrome) —
аутосомно-доминантное заболевание из
Синдром Марфана (Болезнь Марфана, Marfan syndrome) —
аутосомно-доминантное заболевание из

Причина болезни -- мутация в гене, ответственном за синтез белка
Причина болезни -- мутация в гене, ответственном за синтез белка
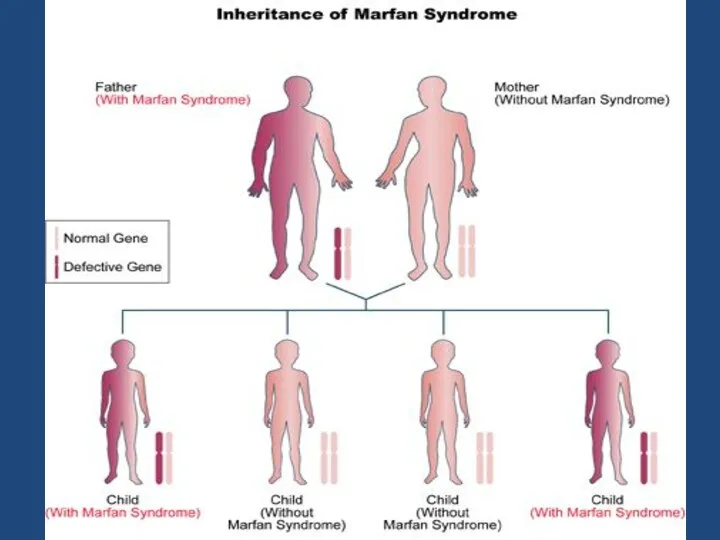
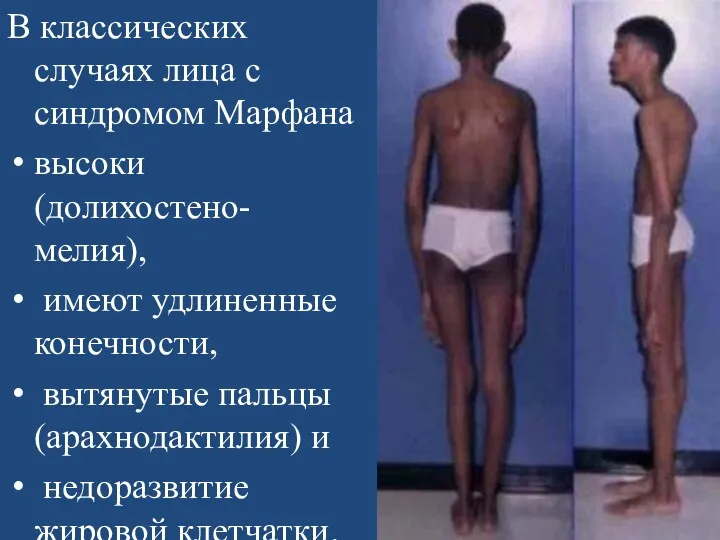
В классических случаях лица с синдромом Марфана
высоки(долихостено-мелия),
имеют удлиненные конечности,
В классических случаях лица с синдромом Марфана
высоки(долихостено-мелия),
имеют удлиненные конечности,

Арахнодактилия
Арахнодактилия
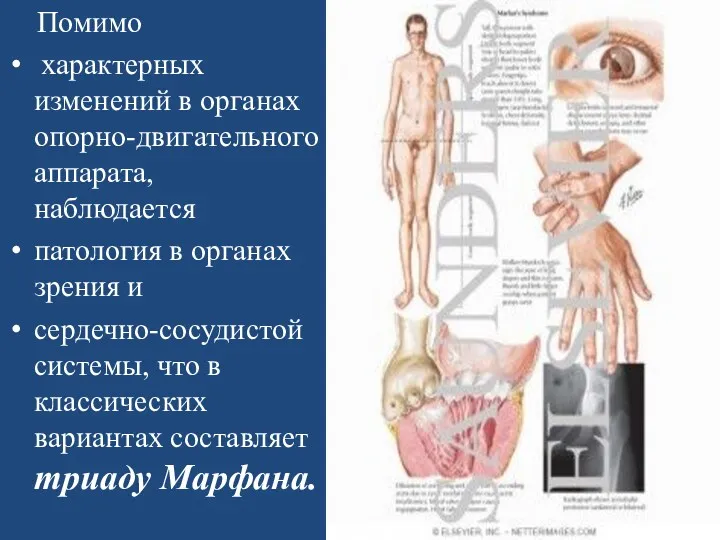
Помимо
характерных изменений в органах опорно-двигательного аппарата, наблюдается
патология в
Помимо
характерных изменений в органах опорно-двигательного аппарата, наблюдается
патология в
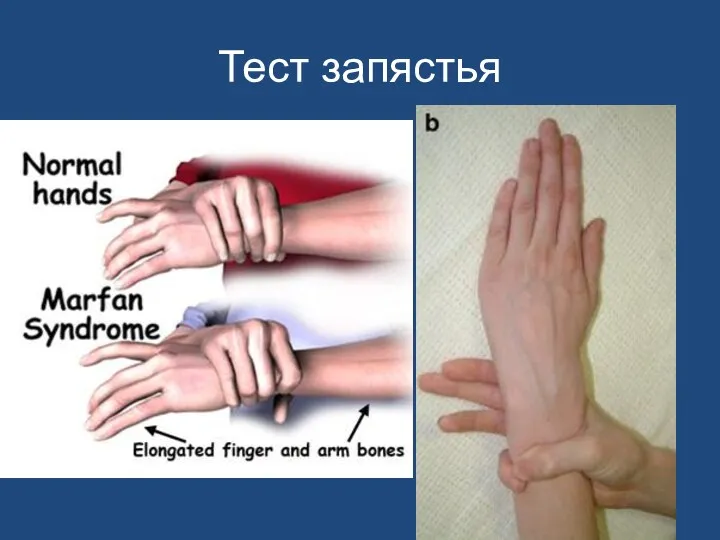
Тест запястья
Тест запястья
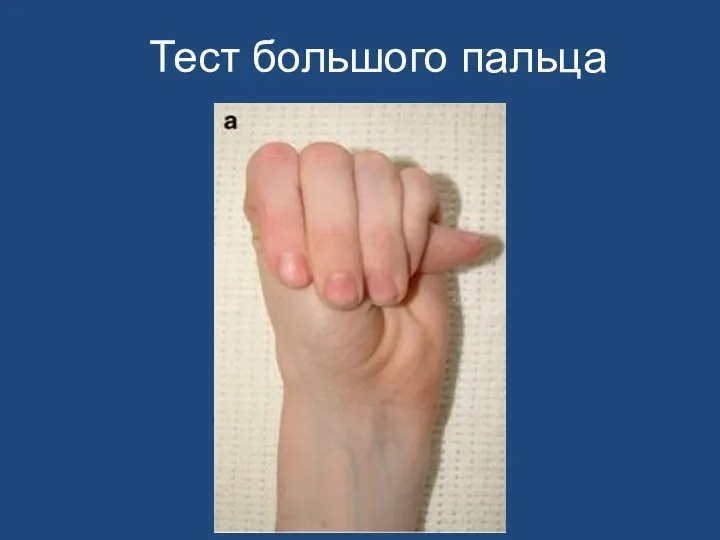
Тест большого пальца
Тест большого пальца
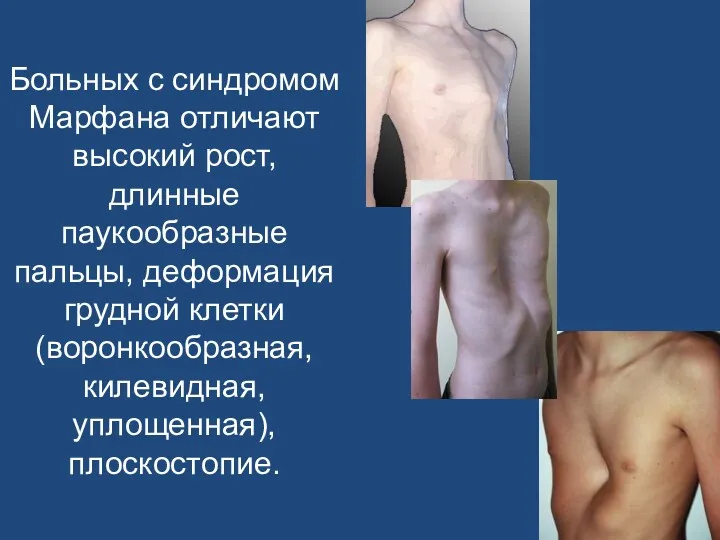
Больных с синдромом Марфана отличают высокий рост, длинные паукообразные пальцы, деформация
Больных с синдромом Марфана отличают высокий рост, длинные паукообразные пальцы, деформация
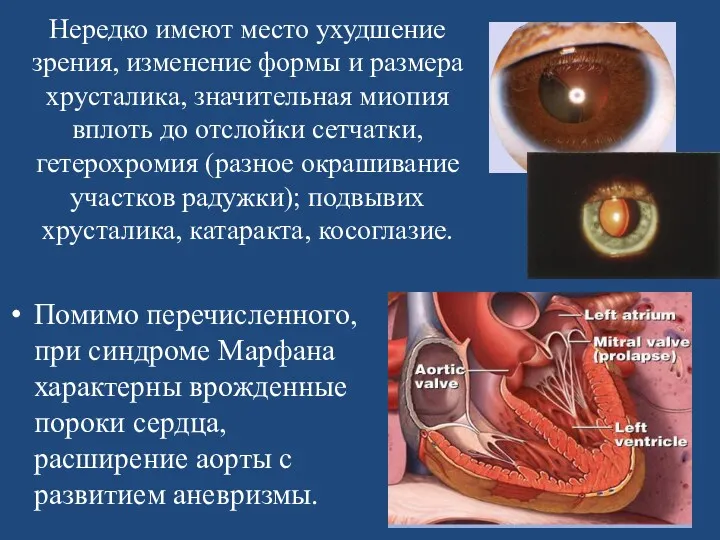
Нередко имеют место ухудшение зрения, изменение формы и размера хрусталика, значительная
Нередко имеют место ухудшение зрения, изменение формы и размера хрусталика, значительная

Лечение в основном симптоматическое. Положительное действие оказывают массаж, лечебная физкультура, а
Лечение в основном симптоматическое. Положительное действие оказывают массаж, лечебная физкультура, а

МУКОПОЛИСАХАРИДОЗ
МУКОПОЛИСАХАРИДОЗ


Мукополисахаридозы
Группа наследственных (генетических) болезней, вызванных аномалиями обмена мукополисахаридов и проявляющихся
Мукополисахаридозы
Группа наследственных (генетических) болезней, вызванных аномалиями обмена мукополисахаридов и проявляющихся

ЭТИОЛОГИЯ
генетический дефект ферментного расщепления углеводной части молекулы мукополисахаридов (гликозоаминогликанов), в тканях
ЭТИОЛОГИЯ
генетический дефект ферментного расщепления углеводной части молекулы мукополисахаридов (гликозоаминогликанов), в тканях

Симптомокомплексы
Нарушается функциональное состояние различных органов и систем, а поскольку гликозаминогликаны входят
Симптомокомплексы
Нарушается функциональное состояние различных органов и систем, а поскольку гликозаминогликаны входят

ТИПЫ
1 ТИП- СИНДРОМ ГУРЛЕР
2 ТИП- СИНДРОМ ХАНТЕРА
3 ТИП- СИНДРОМ САНФИЛИППО
4 ТИП-
ТИПЫ
1 ТИП- СИНДРОМ ГУРЛЕР
2 ТИП- СИНДРОМ ХАНТЕРА
3 ТИП- СИНДРОМ САНФИЛИППО
4 ТИП-

1 ТИП- СИНДРОМ ГУРЛЕР
Голова увеличена
Выражены лобные бугры
Шея почти отсутствует
Рост резко
1 ТИП- СИНДРОМ ГУРЛЕР
Голова увеличена
Выражены лобные бугры
Шея почти отсутствует
Рост резко

Грудная клетка укорочена
Ограничена подвижность в
суставах, контрактуры
Гепатоспленомегалия
Пупочная и паховая грыжи
Склонность к хроническим
ринитам
Шумное
Грудная клетка укорочена
Ограничена подвижность в
суставах, контрактуры
Гепатоспленомегалия
Пупочная и паховая грыжи
Склонность к хроническим
ринитам
Шумное
НЕВРОЛОГИЧЕСКИЙ СТАТУС
гипертензионно-гидроцефальный синдром
диффузная мышечная гипотония
повышение сухожильных рефлексов
общая двигательная заторможенность
снижение интеллекта и
НЕВРОЛОГИЧЕСКИЙ СТАТУС
гипертензионно-гидроцефальный синдром
диффузная мышечная гипотония
повышение сухожильных рефлексов
общая двигательная заторможенность
снижение интеллекта и

ОФТАЛЬМОЛОГИЧЕСКИЕ СИМПТОМЫ
Гипертелоризм
Густые ресницы
Пастозные веки
Макрокорнеа
Конъюнктива век и глазного яблока цианотична, отечна
Утолщение и
ОФТАЛЬМОЛОГИЧЕСКИЕ СИМПТОМЫ
Гипертелоризм
Густые ресницы
Пастозные веки
Макрокорнеа
Конъюнктива век и глазного яблока цианотична, отечна
Утолщение и

СЕРДЕЧНО- СОСУДИСТАЯ СИСТЕМА
Характерны изменения со стороны
- клапанов сердца
- миокарда
-
СЕРДЕЧНО- СОСУДИСТАЯ СИСТЕМА
Характерны изменения со стороны
- клапанов сердца
- миокарда
-
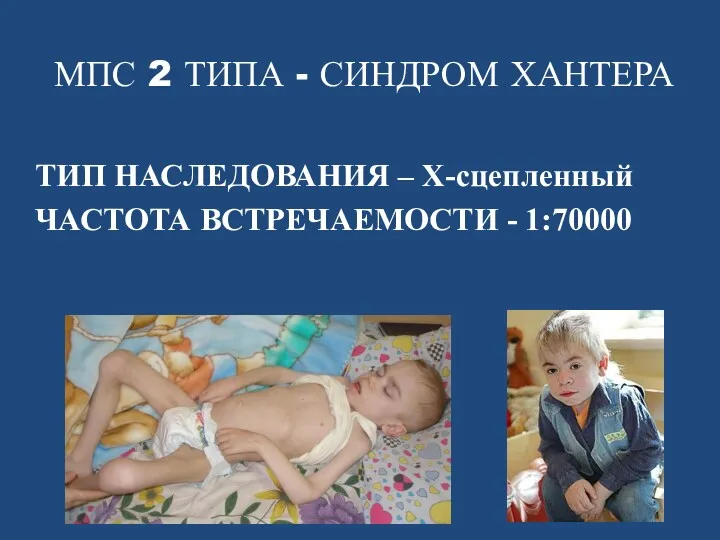
МПС 2 ТИПА - СИНДРОМ ХАНТЕРА
ТИП НАСЛЕДОВАНИЯ – Х-сцепленный
ЧАСТОТА ВСТРЕЧАЕМОСТИ -
МПС 2 ТИПА - СИНДРОМ ХАНТЕРА
ТИП НАСЛЕДОВАНИЯ – Х-сцепленный
ЧАСТОТА ВСТРЕЧАЕМОСТИ -
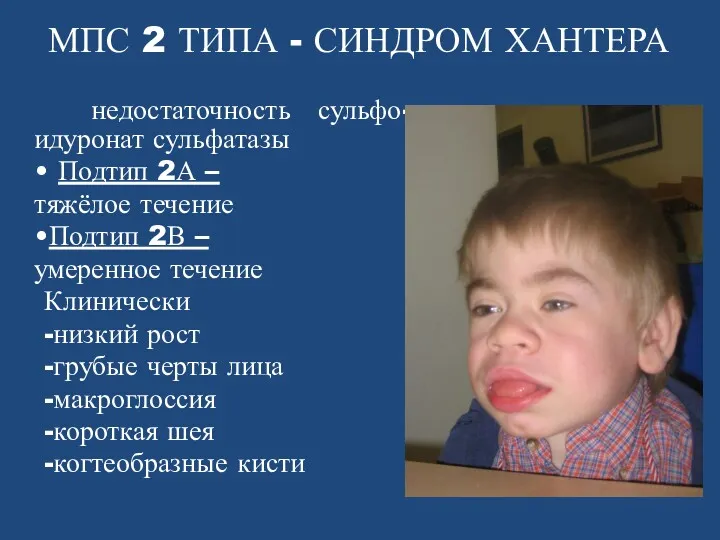
МПС 2 ТИПА - СИНДРОМ ХАНТЕРА
недостаточность сульфо-идуронат сульфатазы
•
МПС 2 ТИПА - СИНДРОМ ХАНТЕРА
недостаточность сульфо-идуронат сульфатазы
•

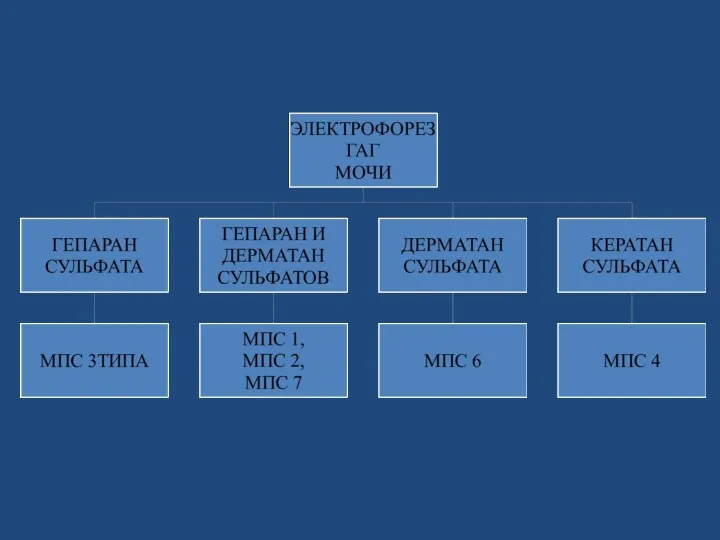
ПРОГРАММА ОБСЛЕДОВАНИЯ БОЛЬНЫХ С МПС
Генеалогический метод
Метод клинического анализа
Рентгено-функциональные методы
Биохимические методы
Морфологические
ПРОГРАММА ОБСЛЕДОВАНИЯ БОЛЬНЫХ С МПС
Генеалогический метод
Метод клинического анализа
Рентгено-функциональные методы
Биохимические методы
Морфологические

КЛИНИЧЕСКАЯ ДИАГНОСТИКА
Выделение главных симптомов и признаков, присущих каждому типу МПС
КЛИНИЧЕСКАЯ ДИАГНОСТИКА
Выделение главных симптомов и признаков, присущих каждому типу МПС

ЛАБОРАТОРНАЯ ДИАГНОСТИКА
Скрининг- тесты (суточная моча на ГАГ – проводится в лаборатории
ЛАБОРАТОРНАЯ ДИАГНОСТИКА
Скрининг- тесты (суточная моча на ГАГ – проводится в лаборатории

ЛЕЧЕНИЕ
1. СИМПТОМАТИЧЕСКАЯ ТЕРАПИЯ
-МЕДИКАМЕНТОЗНАЯ
-ХИРУРГИЧЕСКАЯ
-ФИЗИОТЕРАПЕВТИЧЕСКАЯ
2. ЗАМЕСТИТЕЛЬНАЯ ПАТОГЕНЕТИЧЕСКАЯ ТЕРАПИЯ
-АЛЬДУРАЗИМ
-ЭЛАПРАЗА
ЛЕЧЕНИЕ
1. СИМПТОМАТИЧЕСКАЯ ТЕРАПИЯ
-МЕДИКАМЕНТОЗНАЯ
-ХИРУРГИЧЕСКАЯ
-ФИЗИОТЕРАПЕВТИЧЕСКАЯ
2. ЗАМЕСТИТЕЛЬНАЯ ПАТОГЕНЕТИЧЕСКАЯ ТЕРАПИЯ
-АЛЬДУРАЗИМ
-ЭЛАПРАЗА

ИСХОДЫ
Имеются данные, о том что при легких формах МПС больные могут
ИСХОДЫ
Имеются данные, о том что при легких формах МПС больные могут

ПРОФИЛАКТИКА МПС
Медико-генетическое консультирование семей
Выявление гетерозиготных носителей
Пренатальная диагностика-
определение активности лизосомных ферментов
в
ПРОФИЛАКТИКА МПС
Медико-генетическое консультирование семей
Выявление гетерозиготных носителей
Пренатальная диагностика-
определение активности лизосомных ферментов
в

Наследственные нарушения обмена в эритроцитах
К этой группе относятся болезни, связанные чаще
Наследственные нарушения обмена в эритроцитах
К этой группе относятся болезни, связанные чаще

Гемоглобин -- основной белок эритроцитов. В настоящее время хорошо изучена аминокислотная
Гемоглобин -- основной белок эритроцитов. В настоящее время хорошо изучена аминокислотная

В результате мутаций в эритроцитах и гемоглобине возникают наследственные болезни
В результате мутаций в эритроцитах и гемоглобине возникают наследственные болезни

Гемолитические анемии
включают заболевания, обусловленные снижением уровня гемоглобина и укорочением срока
Гемолитические анемии
включают заболевания, обусловленные снижением уровня гемоглобина и укорочением срока
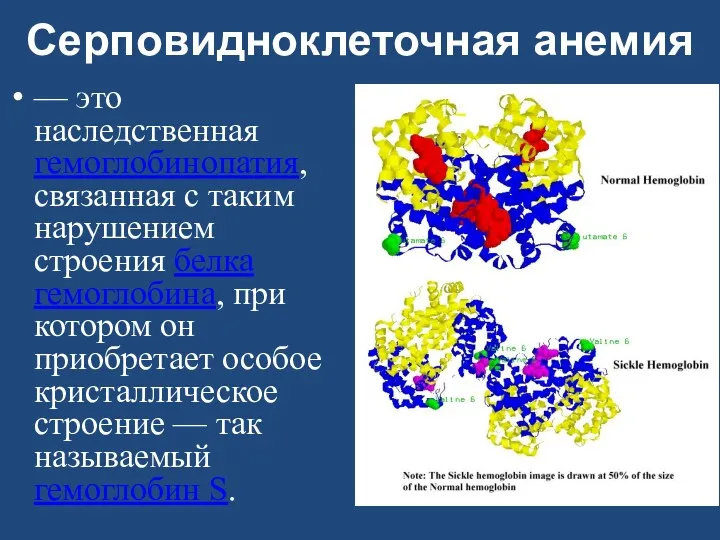
Серповидноклеточная анемия
— это наследственная гемоглобинопатия, связанная с таким нарушением строения
Серповидноклеточная анемия
— это наследственная гемоглобинопатия, связанная с таким нарушением строения
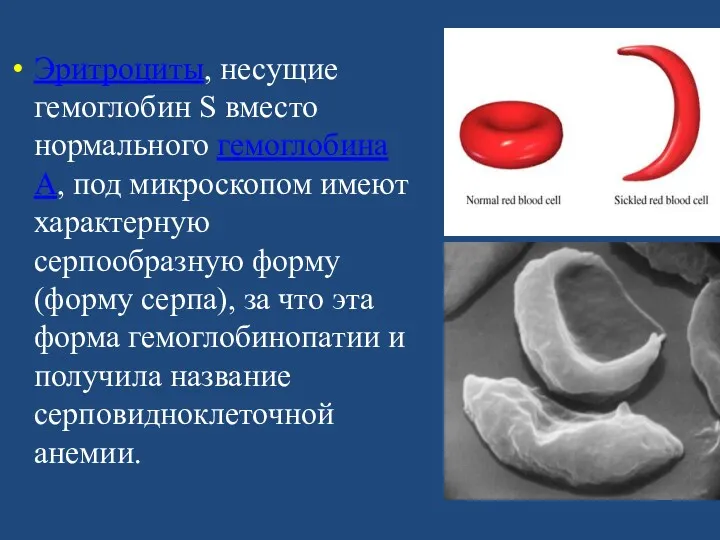
Эритроциты, несущие гемоглобин S вместо нормального гемоглобина А, под микроскопом имеют

Эритроциты, несущие гемоглобин S, обладают пониженной стойкостью и пониженной кислород-транспортирующей способностью,
Эритроциты, несущие гемоглобин S, обладают пониженной стойкостью и пониженной кислород-транспортирующей способностью,
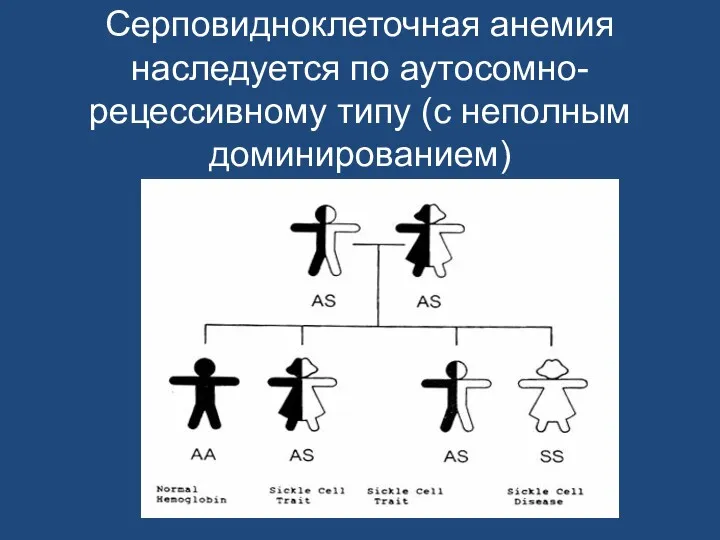
Серповидноклеточная анемия наследуется по аутосомно-рецессивному типу (с неполным доминированием)
Серповидноклеточная анемия наследуется по аутосомно-рецессивному типу (с неполным доминированием)

У носителей, гетерозиготных по гену серповидноклеточной анемии, в эритроцитах присутствуют примерно
У носителей, гетерозиготных по гену серповидноклеточной анемии, в эритроцитах присутствуют примерно

При этом в нормальных условиях у носителей симптомы практически никогда не
При этом в нормальных условиях у носителей симптомы практически никогда не

Симптомы у носителей могут появиться при гипоксии (например, при подъеме в
Симптомы у носителей могут появиться при гипоксии (например, при подъеме в

Серповидноклеточная анемия весьма распространена в регионах мира, эндемичных по малярии, причем
Серповидноклеточная анемия весьма распространена в регионах мира, эндемичных по малярии, причем
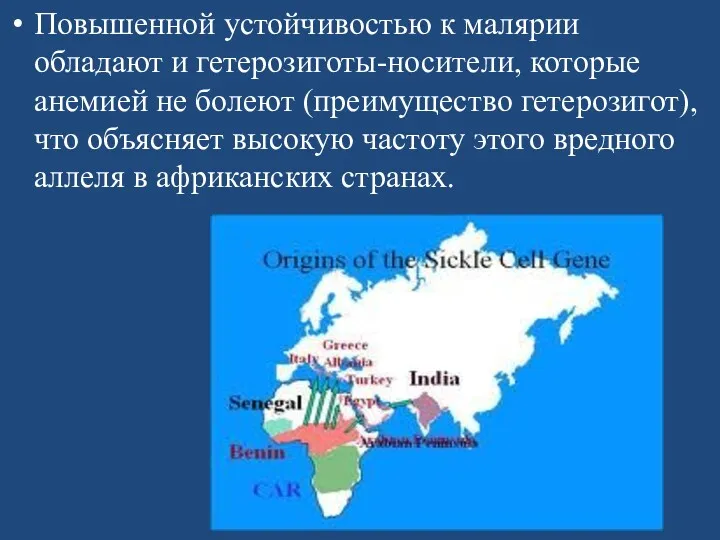
Повышенной устойчивостью к малярии обладают и гетерозиготы-носители, которые анемией не болеют
Повышенной устойчивостью к малярии обладают и гетерозиготы-носители, которые анемией не болеют

Симптомы
Усталость и анемия
Приступы боли
Отек и воспаление пальцев рук и/или ног и
Симптомы
Усталость и анемия
Приступы боли
Отек и воспаление пальцев рук и/или ног и

Симптомы серповидноклеточной анемии делятся на две основные категории.
Из-за хрупкости красных
Симптомы серповидноклеточной анемии делятся на две основные категории.
Из-за хрупкости красных
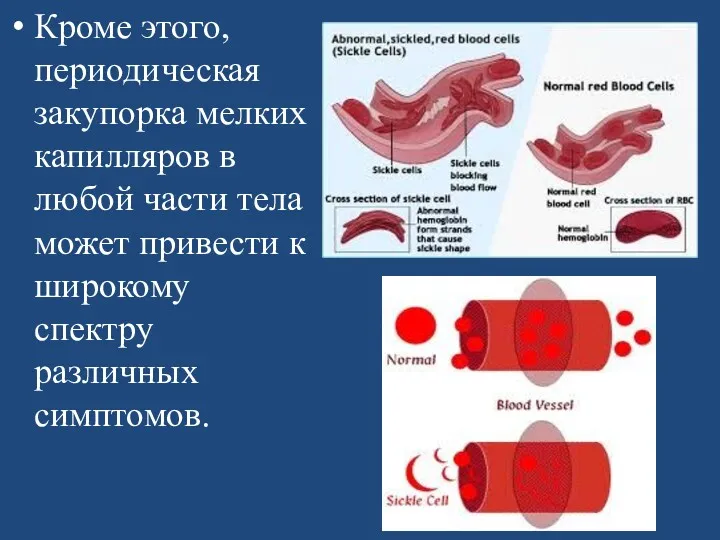
Кроме этого, периодическая закупорка мелких капилляров в любой части тела может
Кроме этого, периодическая закупорка мелких капилляров в любой части тела может
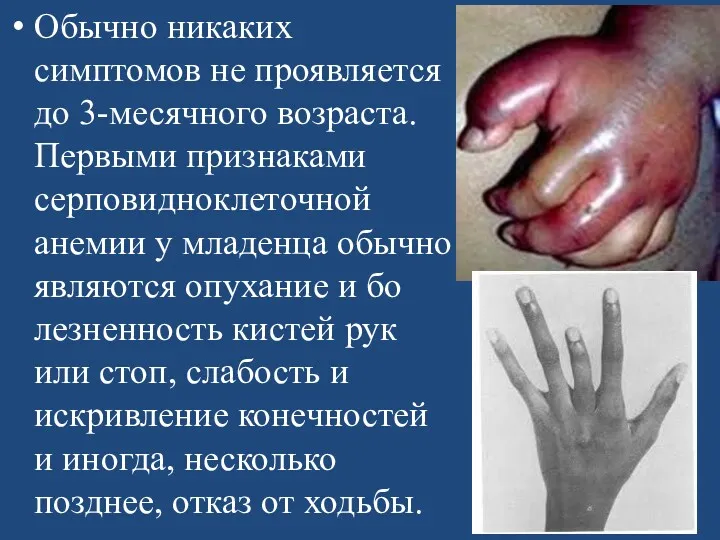
Обычно никаких симптомов не проявляется до 3-месячного возраста. Первыми признаками серповидноклеточной
Обычно никаких симптомов не проявляется до 3-месячного возраста. Первыми признаками серповидноклеточной
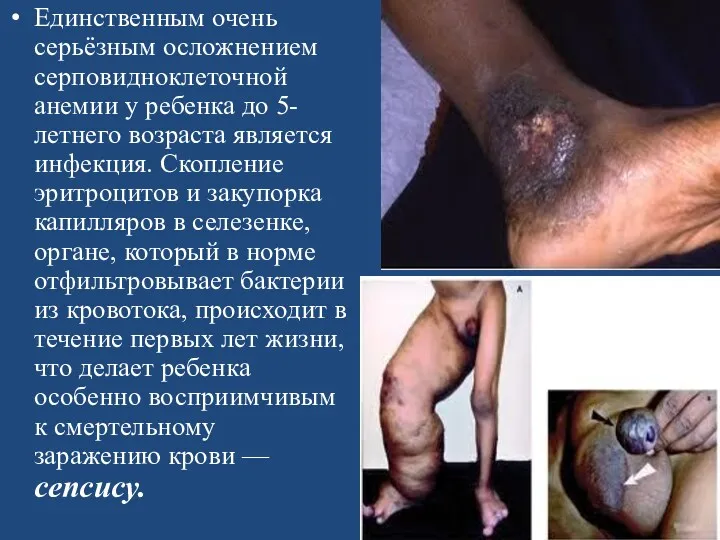
Единственным очень серьёзным осложнением серповидноклеточной анемии у ребенка до 5-летнего возраста
Единственным очень серьёзным осложнением серповидноклеточной анемии у ребенка до 5-летнего возраста
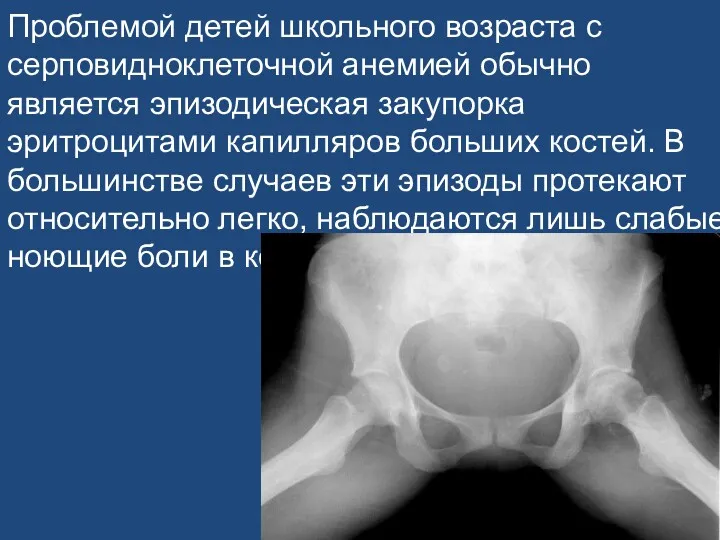
Проблемой детей школьного возраста с серповидноклеточной анемией обычно является эпизодическая закупорка
Проблемой детей школьного возраста с серповидноклеточной анемией обычно является эпизодическая закупорка
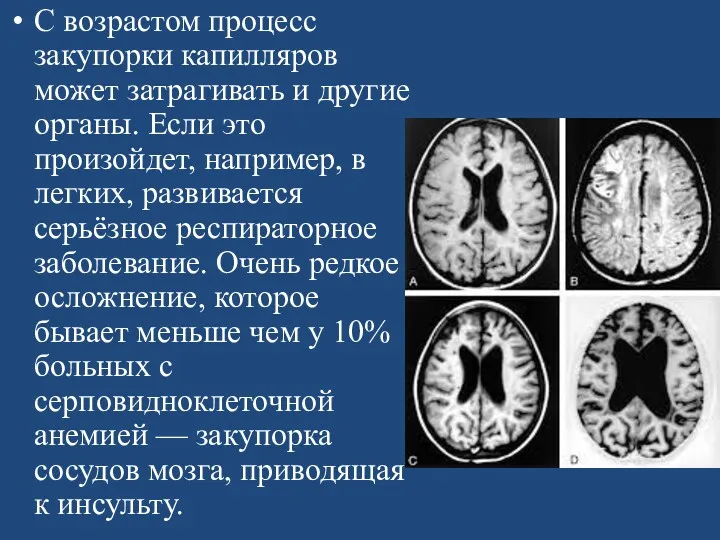
С возрастом процесс закупорки капилляров может затрагивать и другие органы. Если
С возрастом процесс закупорки капилляров может затрагивать и другие органы. Если

У взрослых с серповидноклеточной анемией могут обнаруживаться симптомы хронической (постоянной или
У взрослых с серповидноклеточной анемией могут обнаруживаться симптомы хронической (постоянной или

Специальных методов лечения нет. Важное значение имеет предохранение больного от воздействия
Специальных методов лечения нет. Важное значение имеет предохранение больного от воздействия
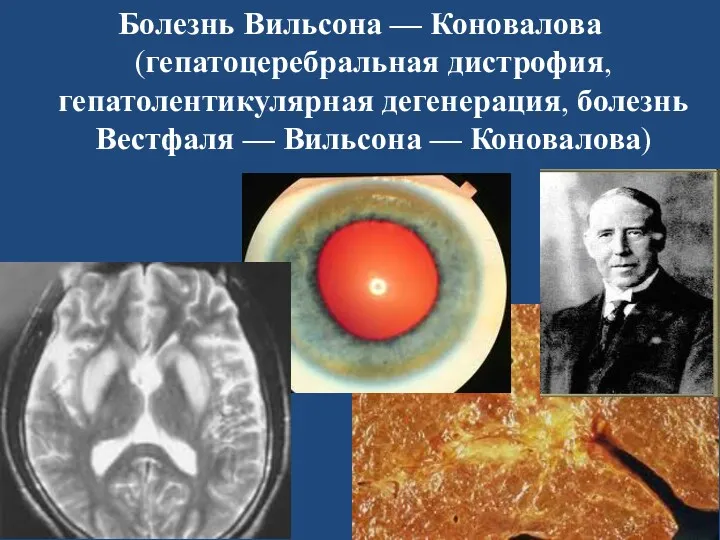
Болезнь Вильсона — Коновалова (гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона — Коновалова)
Болезнь Вильсона — Коновалова (гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона — Коновалова)

Болезнь Вильсона — Коновалова
врождённое нарушение метаболизма меди, приводящее к тяжелейшим наследственным болезням
Болезнь Вильсона — Коновалова
врождённое нарушение метаболизма меди, приводящее к тяжелейшим наследственным болезням
Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста.
Ген
Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста.
Ген

Заболевание передается по аутосомно-рецессивному типу.
Заболевание передается по аутосомно-рецессивному типу.

Основную роль в патогенезе играет нарушение обмена меди, её накопление в
Основную роль в патогенезе играет нарушение обмена меди, её накопление в
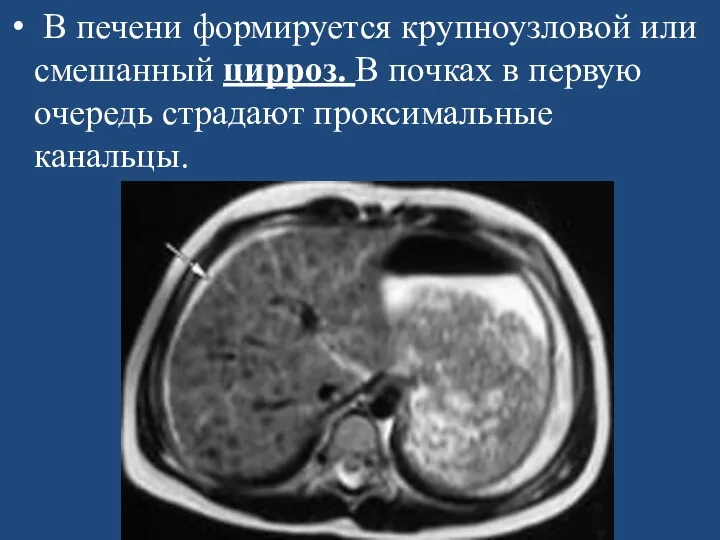
В печени формируется крупноузловой или смешанный цирроз. В почках в
В печени формируется крупноузловой или смешанный цирроз. В почках в
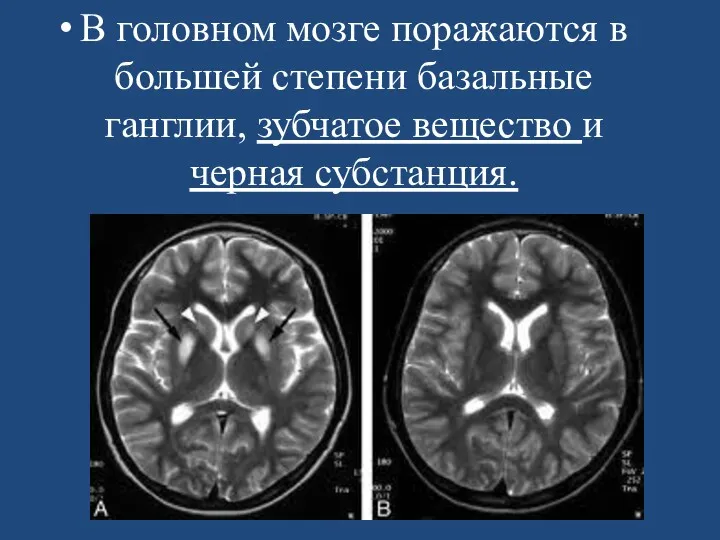
В головном мозге поражаются в большей степени базальные ганглии, зубчатое вещество
В головном мозге поражаются в большей степени базальные ганглии, зубчатое вещество
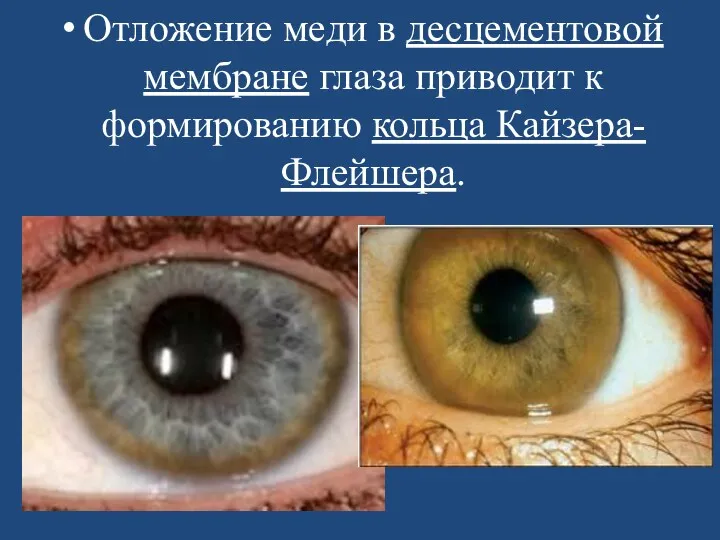
Отложение меди в десцементовой мембране глаза приводит к формированию кольца Кайзера-Флейшера.
Отложение меди в десцементовой мембране глаза приводит к формированию кольца Кайзера-Флейшера.
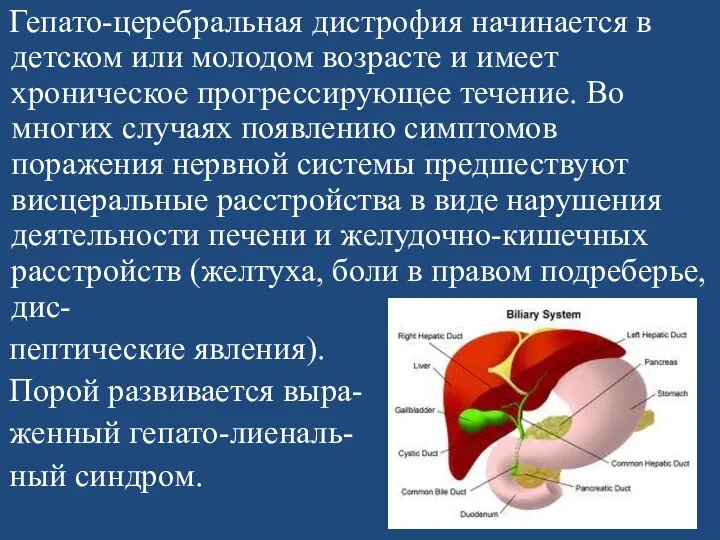
Гепато-церебральная дистрофия начинается в детском или молодом возрасте и имеет
Гепато-церебральная дистрофия начинается в детском или молодом возрасте и имеет

Со стороны нервной системы на первый план выступают экстрапирамидные симптомы в
Со стороны нервной системы на первый план выступают экстрапирамидные симптомы в
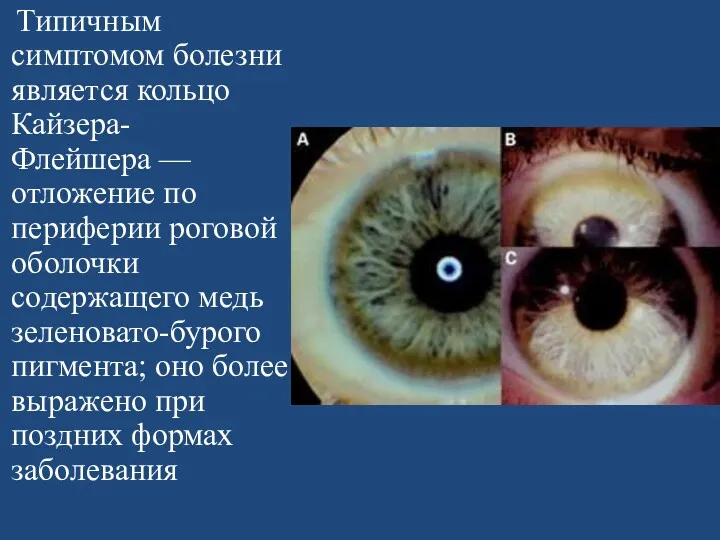
Типичным симптомом болезни является кольцо Кайзера-Флейшера — отложение по периферии роговой
Типичным симптомом болезни является кольцо Кайзера-Флейшера — отложение по периферии роговой
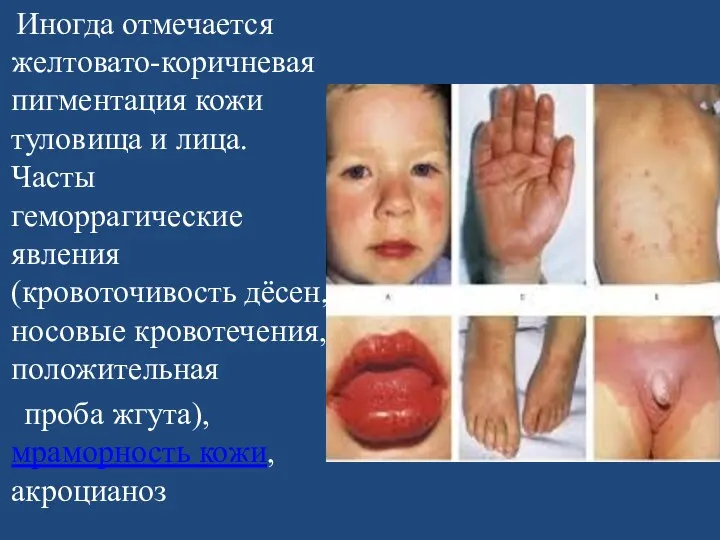
Иногда отмечается желтовато-коричневая пигментация кожи туловища и лица. Часты геморрагические явления

Лечение
Патогенетическое лечение при гепатолентикулярной дегенерации направлено на увеличение выведения меди из
Лечение
Патогенетическое лечение при гепатолентикулярной дегенерации направлено на увеличение выведения меди из

ГЕМОФИЛИЯ
ГЕМОФИЛИЯ

Гемофили́я — наследственное заболевание, связанное с нарушением коагуляции ; при этом
Гемофили́я — наследственное заболевание, связанное с нарушением коагуляции ; при этом

При гемофилии резко возрастает опасность гибели пациента от кровоизлияния в мозг
При гемофилии резко возрастает опасность гибели пациента от кровоизлияния в мозг

Гемофилия появляется из-за изменения одного гена в хромосоме X.
Различают три
Гемофилия появляется из-за изменения одного гена в хромосоме X.
Различают три

Гемофилия A (рецессивная мутация в X-хромосоме) вызывает недостаточность в крови необходимого
Гемофилия A (рецессивная мутация в X-хромосоме) вызывает недостаточность в крови необходимого
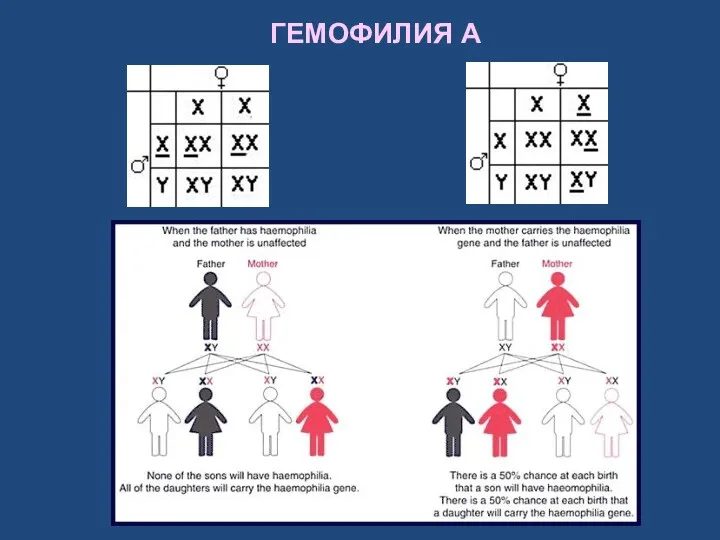
ГЕМОФИЛИЯ A
ГЕМОФИЛИЯ A

Обычно болезнью страдают мужчины (наследование, сцепленное с полом), женщины же обычно
Обычно болезнью страдают мужчины (наследование, сцепленное с полом), женщины же обычно

Общеизвестным является мнение, что женщины не болеют гемофилией, однако это мнение
Общеизвестным является мнение, что женщины не болеют гемофилией, однако это мнение

Кроме того, примерно в 15-25 % случаев обследование матерей мальчиков, страдающих гемофилией,
Кроме того, примерно в 15-25 % случаев обследование матерей мальчиков, страдающих гемофилией,
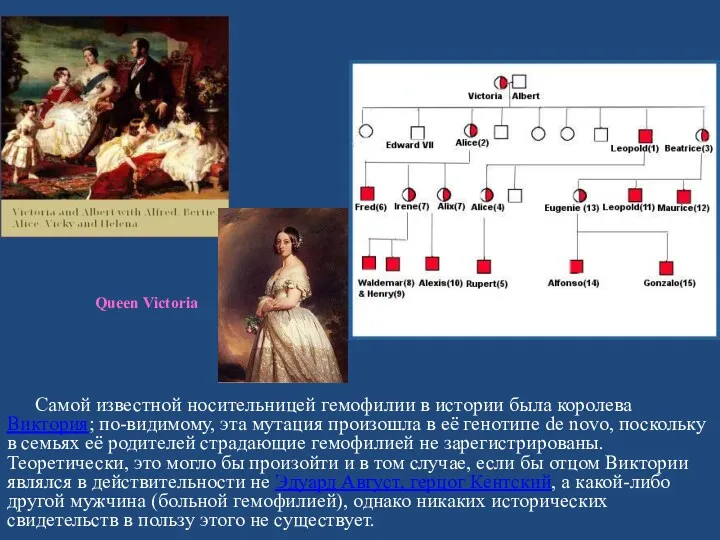
Самой известной носительницей гемофилии в истории была королева Виктория; по-видимому, эта
Самой известной носительницей гемофилии в истории была королева Виктория; по-видимому, эта
Гемофилией страдал один из сыновей Виктории (Леопольд, герцог Олбани), а также
Гемофилией страдал один из сыновей Виктории (Леопольд, герцог Олбани), а также
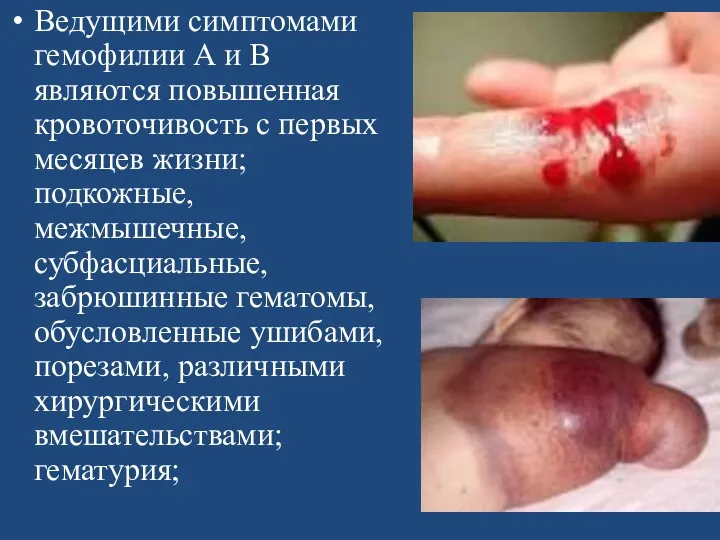
Ведущими симптомами гемофилии А и В являются повышенная кровоточивость с первых
Ведущими симптомами гемофилии А и В являются повышенная кровоточивость с первых
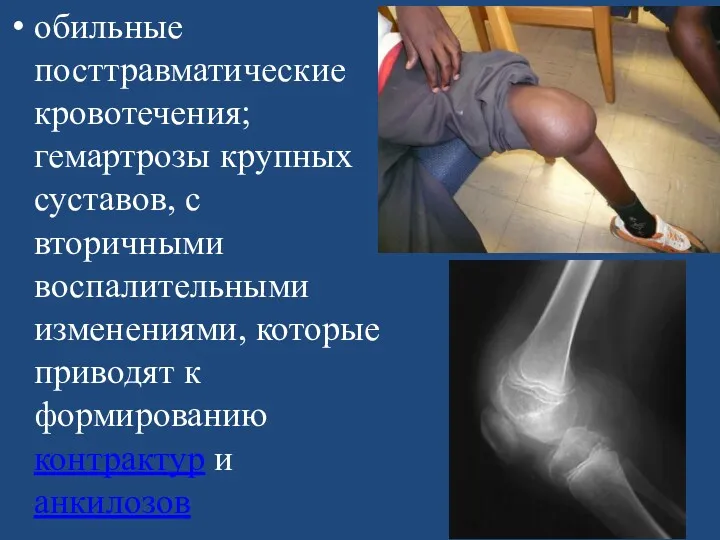
обильные посттравматические кровотечения; гемартрозы крупных суставов, с вторичными воспалительными изменениями, которые
обильные посттравматические кровотечения; гемартрозы крупных суставов, с вторичными воспалительными изменениями, которые
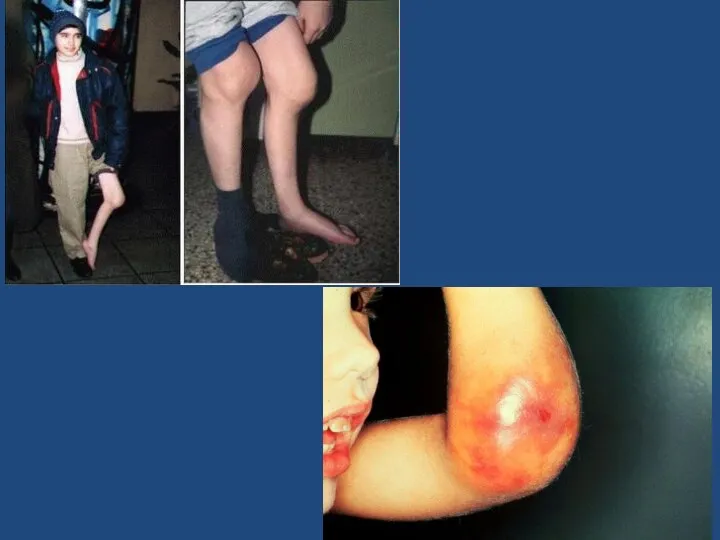

Наиболее распространенное заблуждение о гемофилии — это то, что больной гемофилии может
Наиболее распространенное заблуждение о гемофилии — это то, что больной гемофилии может
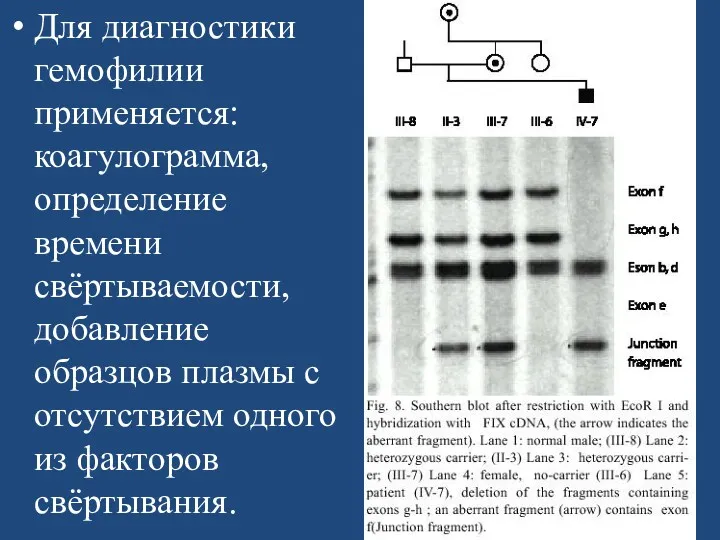
Для диагностики гемофилии применяется: коагулограмма, определение времени свёртываемости, добавление образцов плазмы
Для диагностики гемофилии применяется: коагулограмма, определение времени свёртываемости, добавление образцов плазмы

ЛЕЧЕНИЕ
Хотя болезнь на сегодняшний день неизлечима, её течение контролируется с помощью
ЛЕЧЕНИЕ
Хотя болезнь на сегодняшний день неизлечима, её течение контролируется с помощью

На настоящий момент для лечения используются концентраты факторов свертывания как полученные
На настоящий момент для лечения используются концентраты факторов свертывания как полученные

Гемофилия B (рецессивная мутация в X-хромосоме) недостаточность фактора крови IX (Кристмаса).
Гемофилия B (рецессивная мутация в X-хромосоме) недостаточность фактора крови IX (Кристмаса).
Муковисцидóз (кистозный фиброз)
Муковисцидóз (кистозный фиброз)
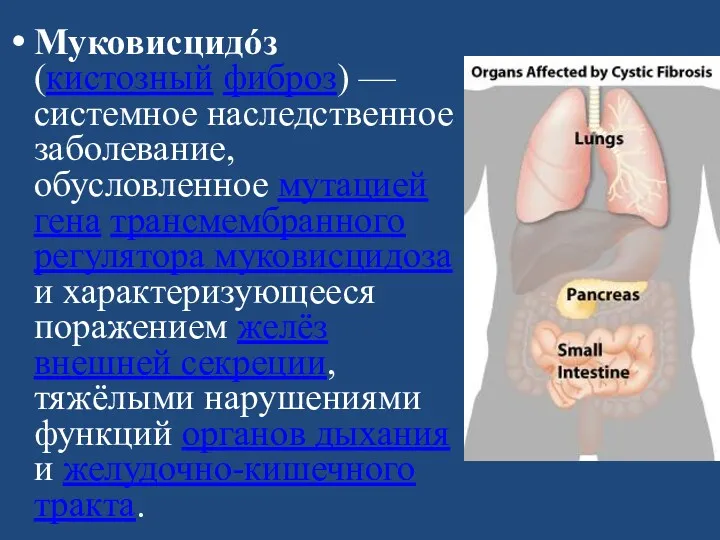
Муковисцидóз (кистозный фиброз) — системное наследственное заболевание, обусловленное мутацией гена трансмембранного регулятора
Муковисцидóз (кистозный фиброз) — системное наследственное заболевание, обусловленное мутацией гена трансмембранного регулятора

Патологический ген локализуется в середине длинного плеча 7-й хромосомы. Муковисцидоз наследуется
Патологический ген локализуется в середине длинного плеча 7-й хромосомы. Муковисцидоз наследуется

Различают следующие клинические формы муковисцидоза:
преимущественно лёгочная форма (респираторная, бронхолёгочная);
преимущественно кишечная форма;
смешанная
Различают следующие клинические формы муковисцидоза:
преимущественно лёгочная форма (респираторная, бронхолёгочная);
преимущественно кишечная форма;
смешанная
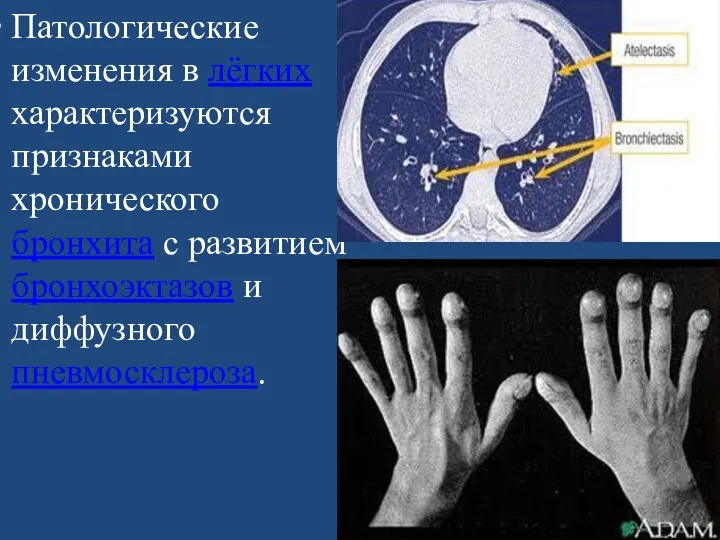
Патологические изменения в лёгких характеризуются признаками хронического бронхита с развитием бронхоэктазов
Патологические изменения в лёгких характеризуются признаками хронического бронхита с развитием бронхоэктазов
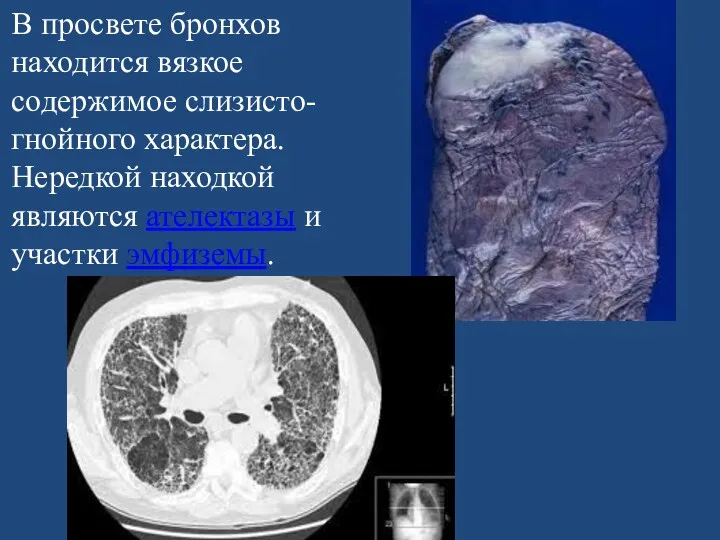
В просвете бронхов находится вязкое содержимое слизисто-гнойного характера. Нередкой находкой являются
В просвете бронхов находится вязкое содержимое слизисто-гнойного характера. Нередкой находкой являются
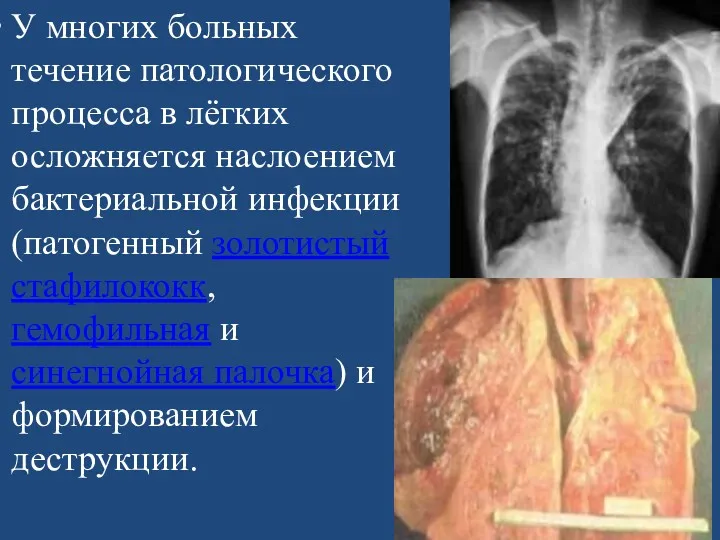
У многих больных течение патологического процесса в лёгких осложняется наслоением бактериальной
У многих больных течение патологического процесса в лёгких осложняется наслоением бактериальной
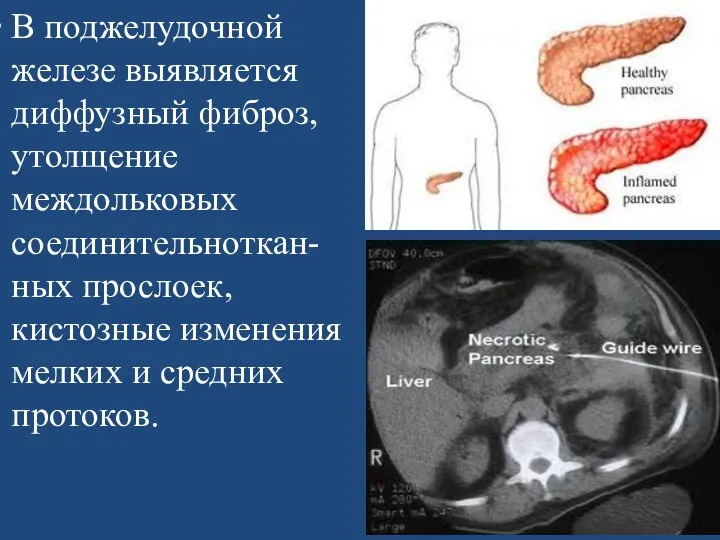
В поджелудочной железе выявляется диффузный фиброз, утолщение междольковых соединительноткан-ных прослоек, кистозные
В поджелудочной железе выявляется диффузный фиброз, утолщение междольковых соединительноткан-ных прослоек, кистозные
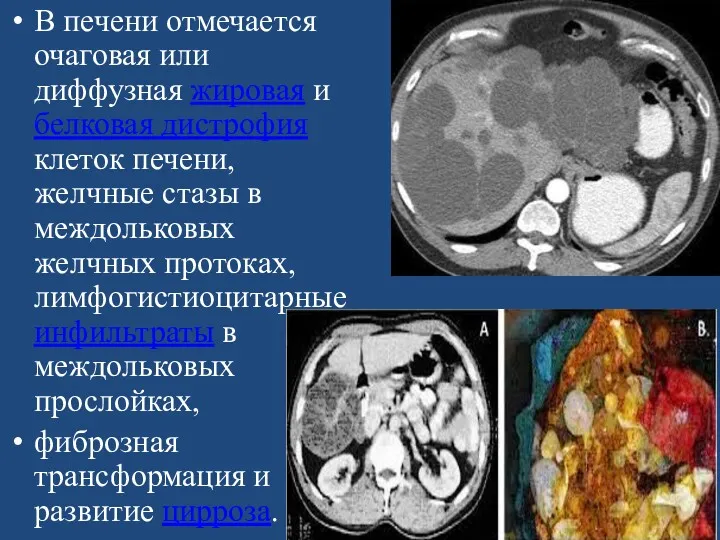
В печени отмечается очаговая или диффузная жировая и белковая дистрофия клеток
В печени отмечается очаговая или диффузная жировая и белковая дистрофия клеток
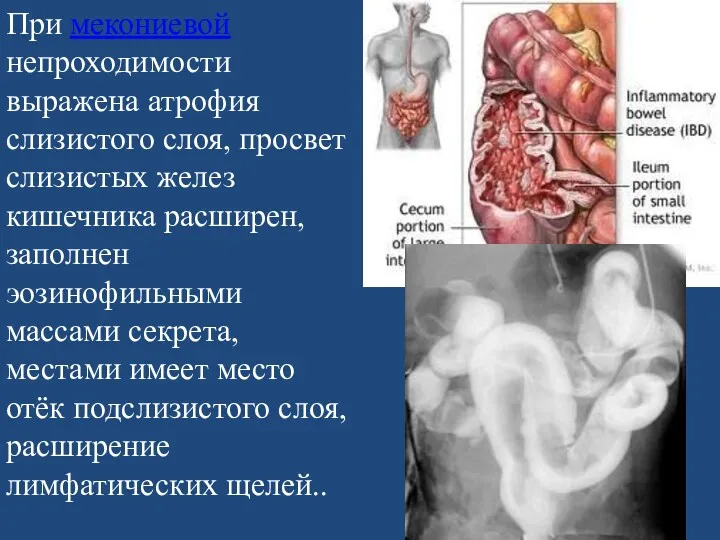
При мекониевой непроходимости выражена атрофия слизистого слоя, просвет слизистых желез кишечника
При мекониевой непроходимости выражена атрофия слизистого слоя, просвет слизистых желез кишечника
Нередко муковисцидоз сочетается с различными пороками развития желудочно-кишечного тракта

Диагностика муковисцидоза
ДНК-диагностика наиболее чувствительная и специфическая. Ложные результаты получают в 0,5—3 %
Диагностика муковисцидоза
ДНК-диагностика наиболее чувствительная и специфическая. Ложные результаты получают в 0,5—3 %

Лечение муковисцидоза
симптоматическое.
коррекция нарушенной функции поджелудочной железы путём применения панкреатина или комбинированных
Лечение муковисцидоза
симптоматическое.
коррекция нарушенной функции поджелудочной железы путём применения панкреатина или комбинированных

Критерием качества диагностики и лечения муковисцидоза является средняя продолжительность жизни больных.
Критерием качества диагностики и лечения муковисцидоза является средняя продолжительность жизни больных.
Нейрофиброматоз
Нейрофиброматоз

Нейрофиброматоз — заболевание из группы факоматозов. Существует 7 типов нейрофиброматоза.
Нейрофиброматоз I
Нейрофиброматоз — заболевание из группы факоматозов. Существует 7 типов нейрофиброматоза.
Нейрофиброматоз I

Нейрофиброматоз II типа (НФ2)
Основными симптомами НФ2 являются:
двусторонняя невринома VIII нерва;
наличие родственника,
Нейрофиброматоз II типа (НФ2)
Основными симптомами НФ2 являются:
двусторонняя невринома VIII нерва;
наличие родственника,

III тип — редкая форма нейрофиброматоза, характеризуется ладонными нейрофибромами, бледноватыми относительно
III тип — редкая форма нейрофиброматоза, характеризуется ладонными нейрофибромами, бледноватыми относительно

Нейрофиброматоз I (первого) типа
( болезнь фон Реклингхаузена, синдром Реклингхаузена, NF-1)
Нейрофиброматоз I (первого) типа ( болезнь фон Реклингхаузена, синдром Реклингхаузена, NF-1)
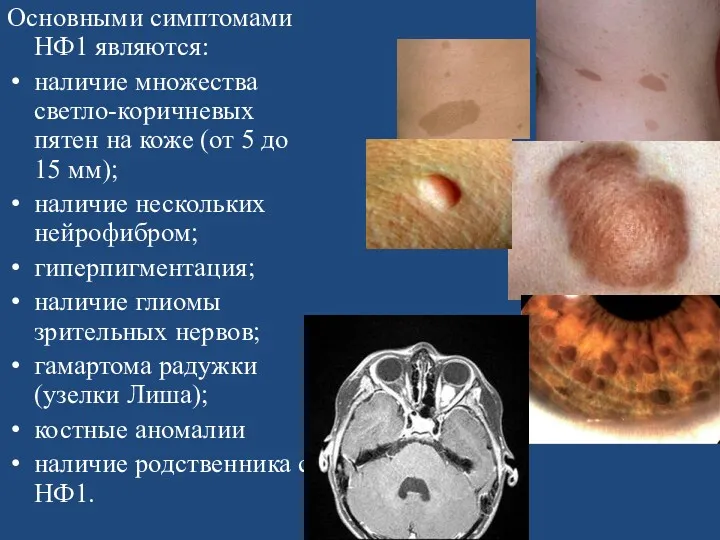
Основными симптомами НФ1 являются:
наличие множества светло-коричневых пятен на коже (от 5
Основными симптомами НФ1 являются:
наличие множества светло-коричневых пятен на коже (от 5
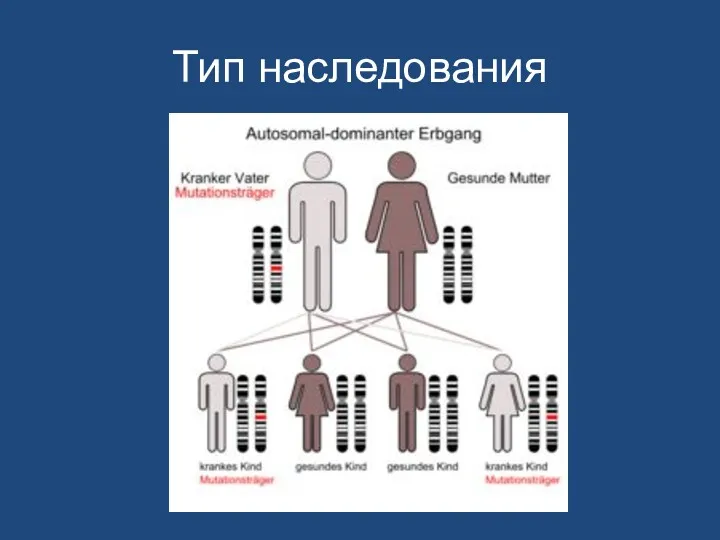
Тип наследования
Тип наследования

Клиническая картина
наличие пигментных пятен на коже цвета «кофе с молоком», нейрофибром,
Клиническая картина
наличие пигментных пятен на коже цвета «кофе с молоком», нейрофибром,
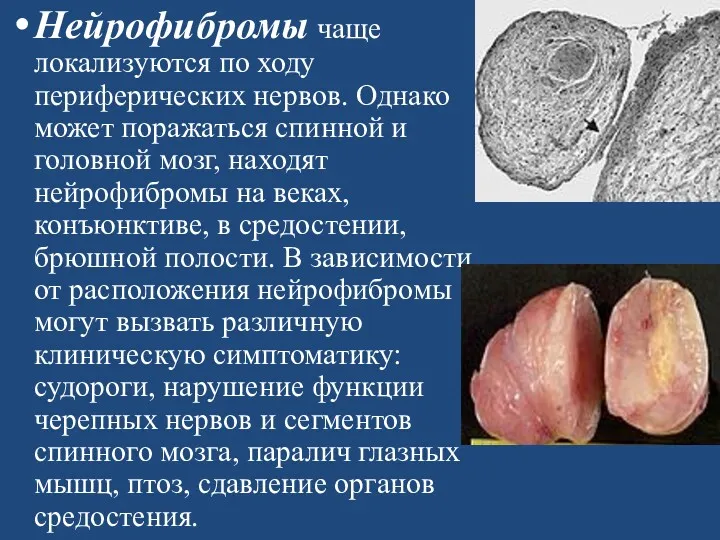
Нейрофибромы чаще локализуются по ходу периферических нервов. Однако может поражаться спинной
Нейрофибромы чаще локализуются по ходу периферических нервов. Однако может поражаться спинной
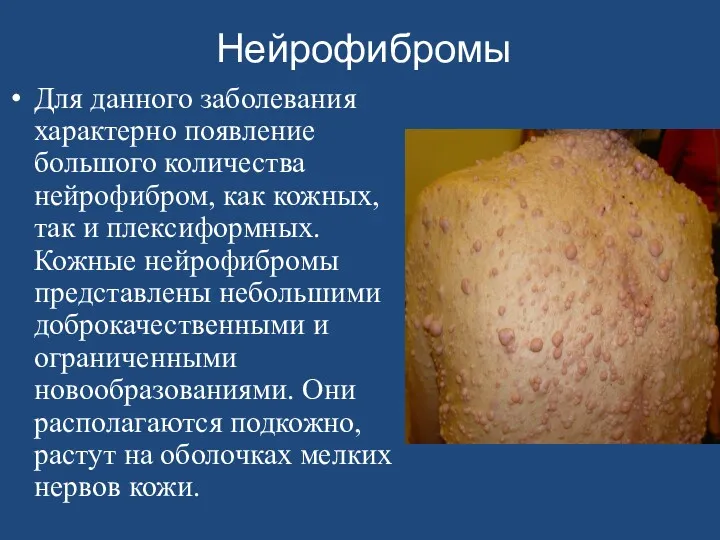
Нейрофибромы
Для данного заболевания характерно появление большого количества нейрофибром, как кожных, так
Нейрофибромы
Для данного заболевания характерно появление большого количества нейрофибром, как кожных, так
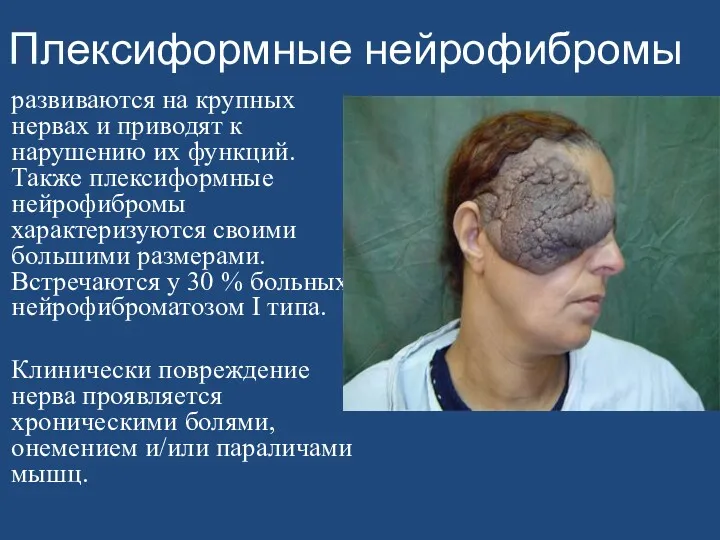
Плексиформные нейрофибромы
развиваются на крупных нервах и приводят к нарушению их функций.
Плексиформные нейрофибромы
развиваются на крупных нервах и приводят к нарушению их функций.
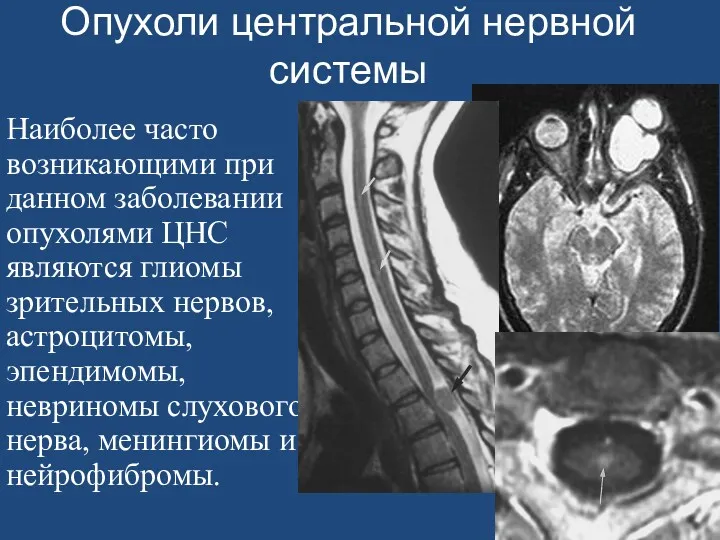
Опухоли центральной нервной системы
Наиболее часто возникающими при данном заболевании опухолями ЦНС
Опухоли центральной нервной системы
Наиболее часто возникающими при данном заболевании опухолями ЦНС
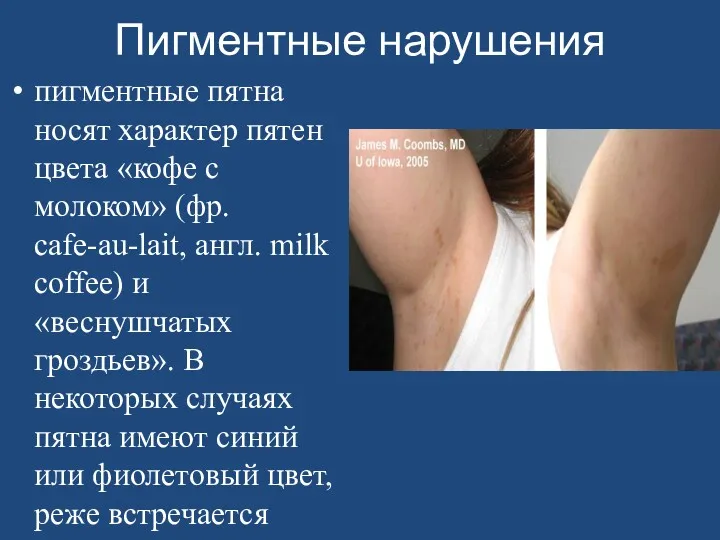
Пигментные нарушения
пигментные пятна носят характер пятен цвета «кофе с молоком» (фр.
Пигментные нарушения
пигментные пятна носят характер пятен цвета «кофе с молоком» (фр.
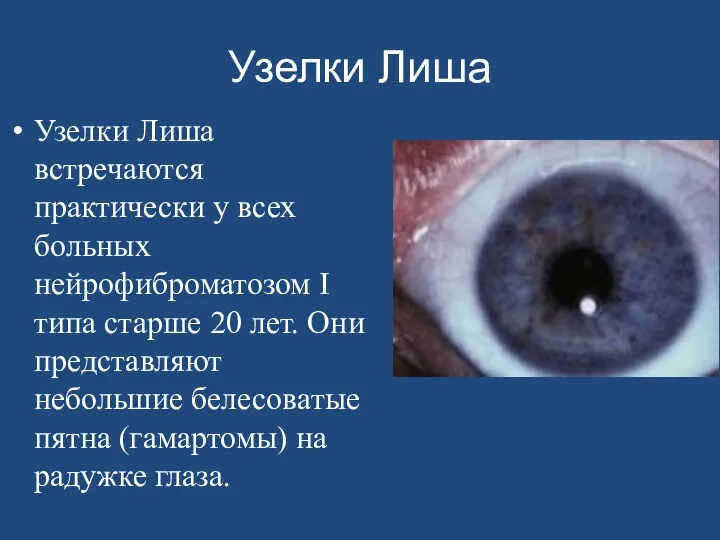
Узелки Лиша
Узелки Лиша встречаются практически у всех больных нейрофиброматозом I типа
Узелки Лиша
Узелки Лиша встречаются практически у всех больных нейрофиброматозом I типа
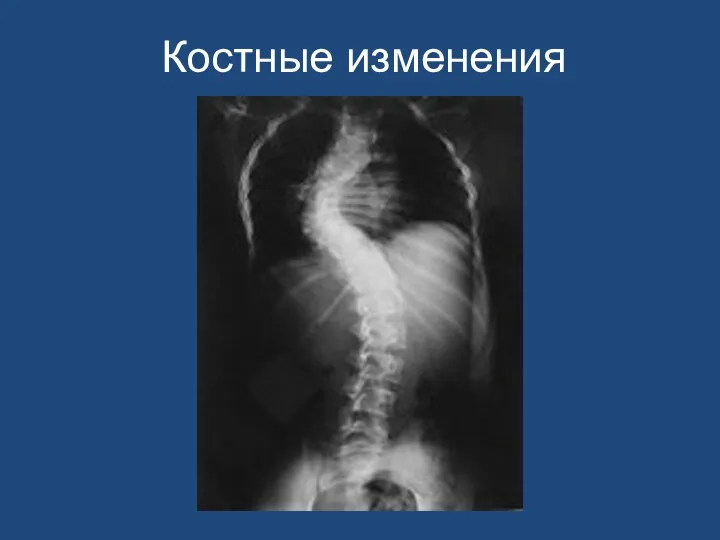
Костные изменения
Костные изменения
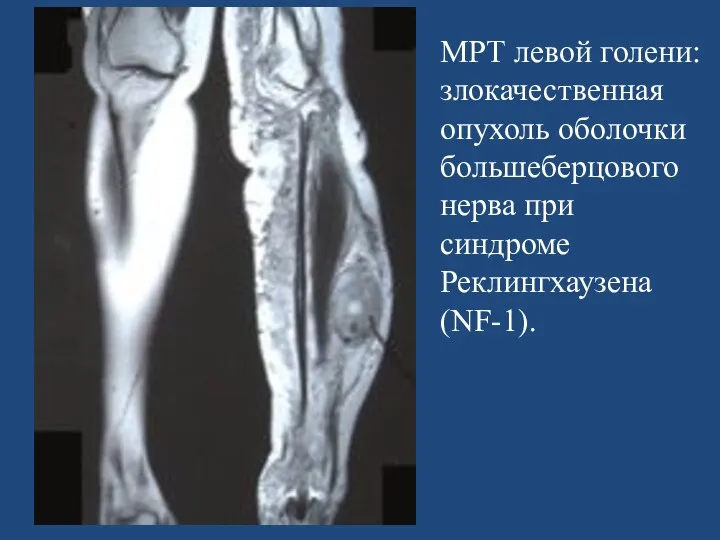
МРТ левой голени: злокачественная опухоль оболочки большеберцового нерва при синдроме Реклингхаузена
МРТ левой голени: злокачественная опухоль оболочки большеберцового нерва при синдроме Реклингхаузена
Лечение
Лечение оперативное. Показаниями для него являются резкая болезненность или изъязвление опухоли,
Лечение
Лечение оперативное. Показаниями для него являются резкая болезненность или изъязвление опухоли,

Нейрофиброматоз II типа
Возникающие при нейрофиброматозе II типа опухоли — доброкачественные, но
Нейрофиброматоз II типа
Возникающие при нейрофиброматозе II типа опухоли — доброкачественные, но
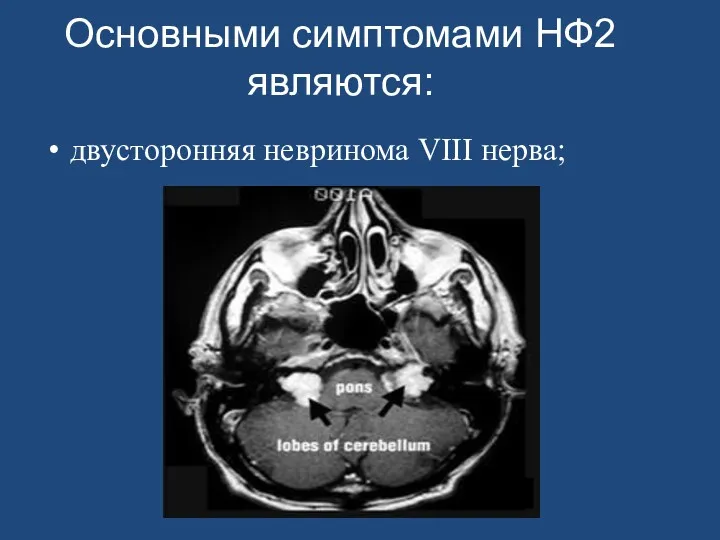
Основными симптомами НФ2 являются:
двусторонняя невринома VIII нерва;
Основными симптомами НФ2 являются:
двусторонняя невринома VIII нерва;
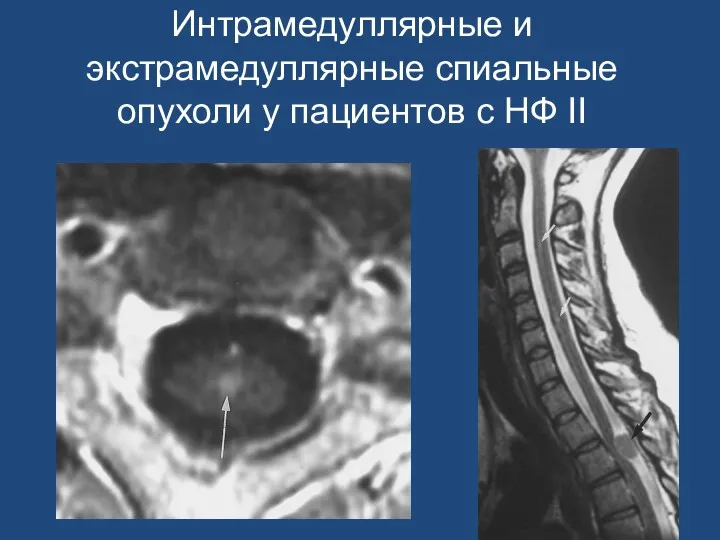
Интрамедуллярные и экстрамедуллярные спиальные опухоли у пациентов с НФ II
Интрамедуллярные и экстрамедуллярные спиальные опухоли у пациентов с НФ II

Лечение
Хирургическое.
При двусторонних невриномах и сохранном слухе, лечение рекомендуется начинать с опухоли
Лечение
Хирургическое.
При двусторонних невриномах и сохранном слухе, лечение рекомендуется начинать с опухоли
 Здоров’я людини і навколишне середовище
Здоров’я людини і навколишне середовище :Остеомиелит
:Остеомиелит Профилактика гриппа и ОРВИ
Профилактика гриппа и ОРВИ Бактериальный вагиноз. Урогенитальный кандидоз
Бактериальный вагиноз. Урогенитальный кандидоз Нетуберкулёзные микобактерии (НТМ). Диагностика и лечение
Нетуберкулёзные микобактерии (НТМ). Диагностика и лечение Нарушение осанки и плоскостопие
Нарушение осанки и плоскостопие Лекарственные средства, действующие на эфферентные нервы. ЛС, действующие в области холинергических синапсов
Лекарственные средства, действующие на эфферентные нервы. ЛС, действующие в области холинергических синапсов Фармацевтические суспензии и эмульсии. Технологические параметры качества фармацевтических субстанций
Фармацевтические суспензии и эмульсии. Технологические параметры качества фармацевтических субстанций Ботулинотерапия в косметологии
Ботулинотерапия в косметологии Итоги работы инфекционной службы Брестской области
Итоги работы инфекционной службы Брестской области ҚР денсаулық сақтаудағы менеджменттің ерекшеліктері мен дамуы
ҚР денсаулық сақтаудағы менеджменттің ерекшеліктері мен дамуы Патофизиология крови (анемии и гемоглобинозы)
Патофизиология крови (анемии и гемоглобинозы) Комалы жагдай шугыл комек
Комалы жагдай шугыл комек Туберкулинді диагностика
Туберкулинді диагностика Жиа. Тұрақты жүктемелік стенокардия
Жиа. Тұрақты жүктемелік стенокардия Роль питания в профилактике онкологических заболеваний
Роль питания в профилактике онкологических заболеваний Противовоспалительные средства
Противовоспалительные средства Основы сердечно-легочной реанимации
Основы сердечно-легочной реанимации Осложнения фармакотерапии противомикробными средствами
Осложнения фармакотерапии противомикробными средствами Патология почек у беременных
Патология почек у беременных Холера – особо опасная инфекция
Холера – особо опасная инфекция Синдром диабетической стопы
Синдром диабетической стопы Наркотики. Быстрый кайф – быстрая смерть
Наркотики. Быстрый кайф – быстрая смерть Способы репаративной регенерации. Понятие регенеративной терапии
Способы репаративной регенерации. Понятие регенеративной терапии Сальмонеллез. Пищевые токсикоинфекции
Сальмонеллез. Пищевые токсикоинфекции Болезнь Бюргера (облитерирующий тромбоангиит)
Болезнь Бюргера (облитерирующий тромбоангиит) Особиста гігієна. Гігієна шкіри, одягу
Особиста гігієна. Гігієна шкіри, одягу Стафилококковые, стрептококковые инфекции, клостридиозы
Стафилококковые, стрептококковые инфекции, клостридиозы