- Мультисистемная атрофия. Оливопонтоцеребеллярная атрофия

Содержание
- 2. Мультисистемная атрофия (МСА) — спорадическое прогрессирующее нейродегенеративное заболевание с поражением базальных ганглиев, ствола мозга, мозжечка, спинного
- 3. КЛАССИФИКАЦИЯ МСА В зависимости от преобладания тех или иных синдромов выделяют 3 основных клинических типа МСА
- 4. КЛАССИФИКАЦИЯ МСА Second consensus statement on the diagnosis of multiple system atrophy. American Autonomic Society and
- 5. Различные клинические варианты МСА описывались в литературе под разными названиями с конца XIX века (Pierret М.,
- 6. РАСПРОСТРАНЕННОСТЬ MSA имеет распространенность 1,9-4,9 случая на 100 000, риск заболеваемости повышается у людей старше 50
- 7. В основе МСА лежит избирательная дегенерация определенных групп нервных и глиальных клеток ЦНС, причины которой остаются
- 8. ПАТОГЕНЕЗ Мультисистемную атрофию можно объяснить как потерю клеток и глиоз или пролиферацию астроцитов в поврежденных участках
- 9. ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ ДЕГЕНЕРАЦИЯ Оливопонтоцеребеллярная дегенерация — заболевание, в основе которого лежат дегенеративные изменения определённых структур мозга —
- 10. ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ Патоморфология Характерными патоморфологическими признаками оливопонтоцеребеллярной дегенерации являются: асимметричная атрофия белого вещества мозжечка, выраженная в
- 11. ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ Клиническая картина Первым симптомом спорадической формы оливопонтоцеребеллярной дегенерации является атактическая походка, чаще появляющаяся в
- 12. ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенераций. I тип (OPCA type
- 13. ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенераций. III тип (OPCA type
- 14. ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенераций. V тип (OPCA type
- 15. ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ ДИАГНОСТИКА Критерии диагноза оливопонтоцеребеллярной дегенерации • Мозжечковая атаксия, дизартрия, экстрапирамидные нарушения, глазодвигательные расстройства, дисфагия,
- 16. Характерен «симптом креста» на аксиальных срезах моста, лучше всего выявляемый в Т2-и диффузионно-взвешенном режимах. Отмеченная картина
- 17. ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ ДИАГНОСТИКА Дифференциальный диагноз оливопонтоцеребеллярной дегенерации проводят с: мозжечковой атаксией различной этиологии, наследственной атаксией Фридрейха,
- 18. Лечение Лечение симптоматическое. Препараты L-DOPA могут на короткое время способствовать уменьшению ригидности и гипокинезии. В случае
- 19. Кафедра Неврологии и Нейрохирургии с курсом последипломного образования Астраханского ГМУ Минздрава России СПАСИБО ЗА ВНИМАНИЕ Ординатор:
- 20. ИСТОЧНИКИ 1. Экстрапирамидные расстройства. Под ред. В.Н. Штока, И.А. Ивановой-Смоленской, О.С. Левина М.: МЕДпресс-информ, 2002. 608
- 22. Скачать презентацию

Мультисистемная атрофия (МСА) — спорадическое прогрессирующее нейродегенеративное заболевание с поражением базальных
Мультисистемная атрофия (МСА) — спорадическое прогрессирующее нейродегенеративное заболевание с поражением базальных

КЛАССИФИКАЦИЯ МСА
В зависимости от преобладания тех или иных синдромов
КЛАССИФИКАЦИЯ МСА
В зависимости от преобладания тех или иных синдромов

КЛАССИФИКАЦИЯ МСА
Second consensus statement on the diagnosis of multiple system
КЛАССИФИКАЦИЯ МСА
Second consensus statement on the diagnosis of multiple system

Различные клинические варианты МСА описывались в литературе под разными названиями
Различные клинические варианты МСА описывались в литературе под разными названиями

РАСПРОСТРАНЕННОСТЬ
MSA имеет распространенность 1,9-4,9 случая на 100 000, риск заболеваемости повышается
РАСПРОСТРАНЕННОСТЬ
MSA имеет распространенность 1,9-4,9 случая на 100 000, риск заболеваемости повышается

В основе МСА лежит избирательная дегенерация определенных групп нервных и
В основе МСА лежит избирательная дегенерация определенных групп нервных и

ПАТОГЕНЕЗ
Мультисистемную атрофию можно объяснить как потерю клеток и глиоз или
ПАТОГЕНЕЗ
Мультисистемную атрофию можно объяснить как потерю клеток и глиоз или

ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ ДЕГЕНЕРАЦИЯ
Оливопонтоцеребеллярная дегенерация — заболевание, в основе которого лежат
ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ ДЕГЕНЕРАЦИЯ
Оливопонтоцеребеллярная дегенерация — заболевание, в основе которого лежат

ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
Патоморфология
Характерными патоморфологическими признаками оливопонтоцеребеллярной дегенерации являются:
асимметричная атрофия
ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
Патоморфология
Характерными патоморфологическими признаками оливопонтоцеребеллярной дегенерации являются:
асимметричная атрофия

ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
Клиническая картина
Первым симптомом спорадической формы оливопонтоцеребеллярной дегенерации является
ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
Клиническая картина
Первым симптомом спорадической формы оливопонтоцеребеллярной дегенерации является

ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенераций.
I
ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенераций.
I

ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенераций.
III
ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенераций.
III

ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенераций.
V
ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенераций.
V

ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
ДИАГНОСТИКА
Критерии диагноза оливопонтоцеребеллярной дегенерации
• Мозжечковая атаксия, дизартрия, экстрапирамидные нарушения, глазодвигательные
ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
ДИАГНОСТИКА
Критерии диагноза оливопонтоцеребеллярной дегенерации
• Мозжечковая атаксия, дизартрия, экстрапирамидные нарушения, глазодвигательные
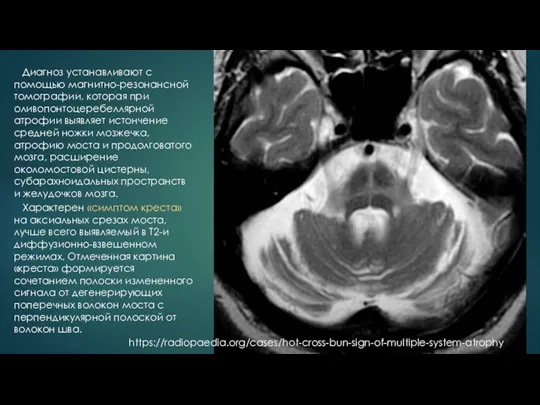
Характерен «симптом креста» на аксиальных срезах моста, лучше всего выявляемый
Характерен «симптом креста» на аксиальных срезах моста, лучше всего выявляемый

ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
ДИАГНОСТИКА
Дифференциальный диагноз оливопонтоцеребеллярной дегенерации проводят с:
мозжечковой атаксией различной
ОЛИВОПОНТОЦЕРЕБЕЛЛЯРНАЯ АТРОФИЯ
ДИАГНОСТИКА
Дифференциальный диагноз оливопонтоцеребеллярной дегенерации проводят с:
мозжечковой атаксией различной

Лечение
Лечение симптоматическое.
Препараты L-DOPA могут на короткое время способствовать уменьшению
Лечение
Лечение симптоматическое.
Препараты L-DOPA могут на короткое время способствовать уменьшению

Кафедра Неврологии и Нейрохирургии
с курсом последипломного образования
Астраханского ГМУ Минздрава России
СПАСИБО ЗА
Кафедра Неврологии и Нейрохирургии
с курсом последипломного образования
Астраханского ГМУ Минздрава России
СПАСИБО ЗА

ИСТОЧНИКИ
1. Экстрапирамидные расстройства. Под ред. В.Н. Штока, И.А. Ивановой-Смоленской, О.С. Левина
М.:
ИСТОЧНИКИ
1. Экстрапирамидные расстройства. Под ред. В.Н. Штока, И.А. Ивановой-Смоленской, О.С. Левина
М.:
 Общение в сестринском деле
Общение в сестринском деле Гигиена питания
Гигиена питания Алгоритм диагностики и оказания скорой помощи при шоковых состояниях
Алгоритм диагностики и оказания скорой помощи при шоковых состояниях Диабетическая офтальмопатия
Диабетическая офтальмопатия Кишечные инфекции: ОКИ, микотические и паразитарные поражения кишечника
Кишечные инфекции: ОКИ, микотические и паразитарные поражения кишечника Травма живота у детей. Тактика лечения. Травматический разрыв печени
Травма живота у детей. Тактика лечения. Травматический разрыв печени Организация и структура системы первичной медико-санитарной помощи
Организация и структура системы первичной медико-санитарной помощи Участие медицинской сестры в лабораторных исследованиях пациента
Участие медицинской сестры в лабораторных исследованиях пациента Аллергия. Иммунопатологические реакции
Аллергия. Иммунопатологические реакции Снятие швов с послеоперационной раны
Снятие швов с послеоперационной раны Ошибки диагностики и лечения при сочетанной травме
Ошибки диагностики и лечения при сочетанной травме Ароматерапия в жизни человека
Ароматерапия в жизни человека Лучевая болезнь
Лучевая болезнь Пародонтоз. Клиникасы, диагностикасы, дифференциальды диагностикасы, пародонтоздың патологиялық анатомиясы
Пародонтоз. Клиникасы, диагностикасы, дифференциальды диагностикасы, пародонтоздың патологиялық анатомиясы Сучасні проблеми молекулярної біології. Генна терапія. (Лекція 8)
Сучасні проблеми молекулярної біології. Генна терапія. (Лекція 8) Гипоксически-ишемические поражения головного мозга у новорожденных
Гипоксически-ишемические поражения головного мозга у новорожденных Особенности анестезиологии и интенсивной терапии в педиатрической практике
Особенности анестезиологии и интенсивной терапии в педиатрической практике Фортранс. Рекомендации по приёму препарата фортранс
Фортранс. Рекомендации по приёму препарата фортранс Ультразвук для терапии боли
Ультразвук для терапии боли Клинический случай
Клинический случай Логоритмика как средство коррекции речевых и неречевых нарушений
Логоритмика как средство коррекции речевых и неречевых нарушений Клинические проявления бронхиальной астмы
Клинические проявления бронхиальной астмы Ожирение как глобальная проблема человечества
Ожирение как глобальная проблема человечества Зубы и уход за ними
Зубы и уход за ними Дыхательная гимнастика для детей дошкольного возраста
Дыхательная гимнастика для детей дошкольного возраста Влияние радиации на здоровье человека
Влияние радиации на здоровье человека Современные подходы в лечении сахарного диабета 2 типа
Современные подходы в лечении сахарного диабета 2 типа Влияние компьютера на здоровье человека
Влияние компьютера на здоровье человека