- Наследственные болезни обмена

Содержание
- 2. Наследственные болезни обмена – группа моногенных наследственных заболеваний, обусловленных мутациями генов, кодирующих ферменты, транспортные или сигнальные
- 3. МАР АНТИМОНГОЛОИДНЫЙ РАЗРЕЗ ГЛАЗ
- 4. ТЕЛЕКАНТ ЭПИКАНТ КАРПИЙ РОТ
- 5. Характерные симптомы 1. Наличие светлого промежутка 2. Симптомокомплекс «вялого ребенка» 3. Мышечная гипотония, сменяющаяся дистонией 4.
- 6. Фенилкетонурия Накопление Фенилаланина и его токсическое воздействие Мышиный запах мочи Дети светловолосые, голубоглазые Светочувствительность (дерматозы)
- 7. Лейциноз Снижение активности энзимной системы, обеспечивающей декарбоксилирование трех аминокислот Моча с запахом кленового сиропа
- 8. Вильсона-Коновалова Нарушение структуры медьтранспортирующей АТФазы печени Токсическое влияние меди на ГМ, почки, печень, роговицу. Патогенетическое лечение
- 9. Гемохроматоз Гепатомегалия меланодермия сахарный диабет миокардиопатия гипогонадизм у взрослых артропатии синдром мальабсорбции. Есть патогенетическое лечение –
- 10. Х-сцепленная адренолейкодистрофия Нарушение транспорта ОДЦЖК в пероксисомы, нарушение расщепления ОДЦЖК Токсическое воздействие на миелин и клетки
- 11. Лизосомные болезни обмена Болезнь Нимана-Пика Мукополисахаридоз Болезнь Гоше
- 12. Болезнь Нимана-Пика Накопление сфингомиелина в лизосомах ГМ, печени, РЭС. Пенистые клетки Симптом «вишневой косточки»
- 13. Тип С Ювенильная форма + Филиппиновый тест
- 14. Мукополисахаридоз Накопление мукополисахаридов в результате дефекта различных ферментов МПС 1 Гурлера Шейе Гурлер-Шейе Дефицит альфа-L-идуронидазы МПС
- 15. МПС
- 16. МПС 1 типа Синдром Гурлера Синдром Шейе Синдром Гурлера-Шейе
- 17. МПС 2 Синдром Хантера Светлы жесткие волосы Узелково-папулезное поражение кожи К 8 годам – умственная отсталость
- 18. МПС III Синдром Санфилиппо К 3 годам жизни развитие прекращается Физическое развитие соответствует возрасту Расстройства поведения
- 19. МПС IV Болезнь Моркио Отсутствие снижения интеллекта Деформация скелета Глухота
- 20. МПС VI Синдром Марото-Лами Отсутствие снижения интеллекта Тугоподвижность суставов Рестриктивные заболевания легких Дисплазия головки бедренной кости
- 21. Болезнь Гоше Накопление глюкоцереброзида Гепатоспленомегалия Для 1 и 3 типа – заместительная терапия Церезин*
- 22. БГ тип 1 до и после ферментзаместительной терапии Девочка 11 мес, БГ тип 2 Мальчик 3
- 23. Митохондриальные заболевания Феномен гетероплазии – присутствие мутантных и нормальных мтДНК в одной клектке Клиническая картина зависит
- 24. Синдром Кернса-Сейра Наружная офтальмоплегия Миопатия Атаксия Нейросенсорная глухота Феномен «рваных красных волокон» в мышечных биоптатах
- 26. Скачать презентацию

Наследственные болезни обмена
– группа моногенных наследственных заболеваний, обусловленных мутациями генов,
Наследственные болезни обмена
– группа моногенных наследственных заболеваний, обусловленных мутациями генов,
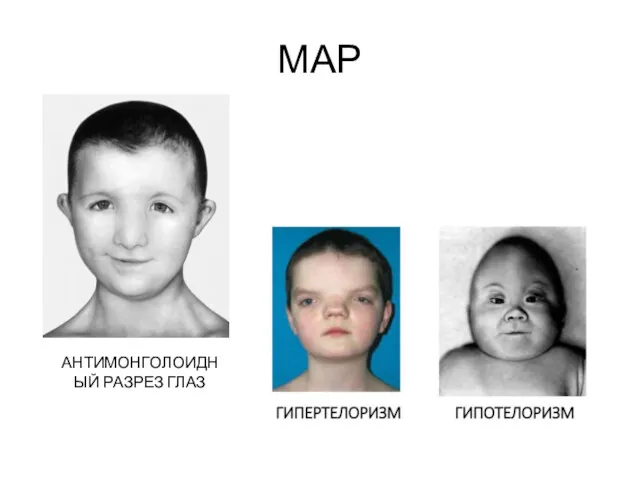
МАР
АНТИМОНГОЛОИДНЫЙ РАЗРЕЗ ГЛАЗ
МАР
АНТИМОНГОЛОИДНЫЙ РАЗРЕЗ ГЛАЗ
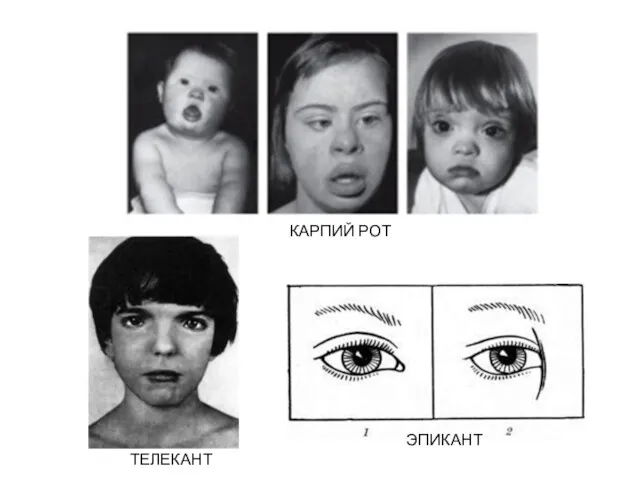
ТЕЛЕКАНТ
ЭПИКАНТ
КАРПИЙ РОТ
ТЕЛЕКАНТ
ЭПИКАНТ
КАРПИЙ РОТ

Характерные симптомы
1. Наличие светлого промежутка
2. Симптомокомплекс «вялого ребенка»
3. Мышечная гипотония, сменяющаяся
Характерные симптомы
1. Наличие светлого промежутка
2. Симптомокомплекс «вялого ребенка»
3. Мышечная гипотония, сменяющаяся

Фенилкетонурия
Накопление Фенилаланина и его токсическое воздействие
Мышиный запах мочи
Дети светловолосые, голубоглазые
Светочувствительность (дерматозы)
Фенилкетонурия
Накопление Фенилаланина и его токсическое воздействие
Мышиный запах мочи
Дети светловолосые, голубоглазые
Светочувствительность (дерматозы)
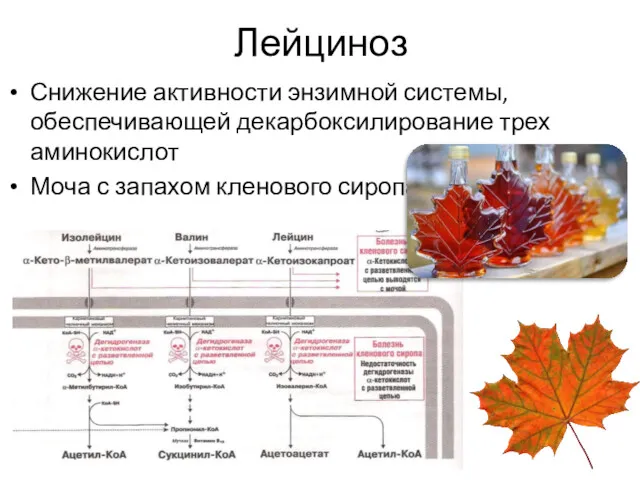
Лейциноз
Снижение активности энзимной системы, обеспечивающей декарбоксилирование трех аминокислот
Моча с запахом кленового
Лейциноз
Снижение активности энзимной системы, обеспечивающей декарбоксилирование трех аминокислот
Моча с запахом кленового
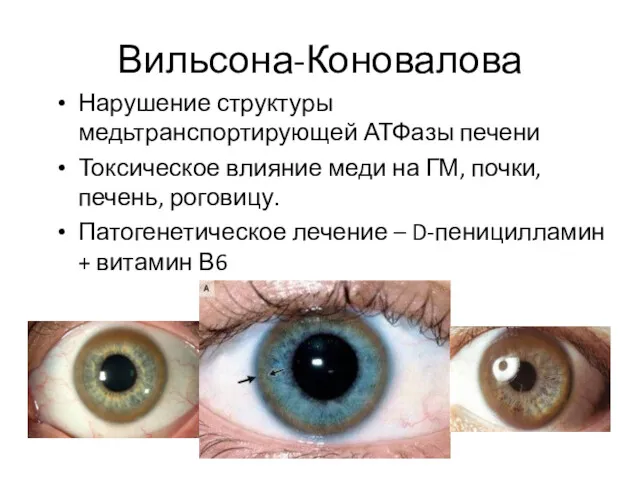
Вильсона-Коновалова
Нарушение структуры медьтранспортирующей АТФазы печени
Токсическое влияние меди на ГМ, почки, печень,
Вильсона-Коновалова
Нарушение структуры медьтранспортирующей АТФазы печени
Токсическое влияние меди на ГМ, почки, печень,

Гемохроматоз
Гепатомегалия меланодермия сахарный диабет миокардиопатия гипогонадизм у взрослых артропатии синдром мальабсорбции.
Есть
Гемохроматоз
Гепатомегалия меланодермия сахарный диабет миокардиопатия гипогонадизм у взрослых артропатии синдром мальабсорбции.
Есть
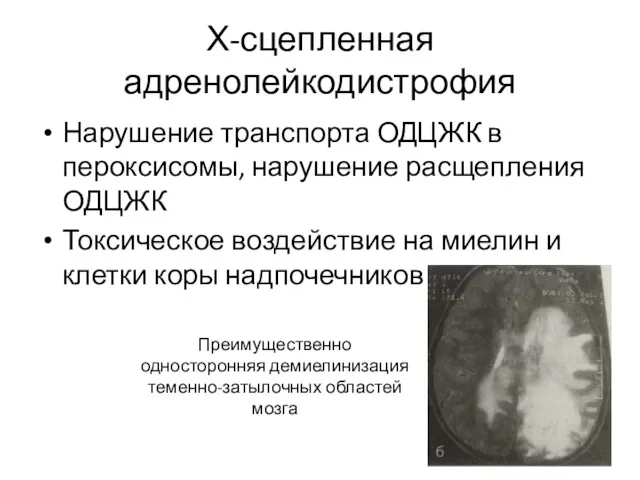
Х-сцепленная адренолейкодистрофия
Нарушение транспорта ОДЦЖК в пероксисомы, нарушение расщепления ОДЦЖК
Токсическое воздействие на
Х-сцепленная адренолейкодистрофия
Нарушение транспорта ОДЦЖК в пероксисомы, нарушение расщепления ОДЦЖК
Токсическое воздействие на
Лизосомные болезни обмена
Болезнь Нимана-Пика
Мукополисахаридоз
Болезнь Гоше
Лизосомные болезни обмена
Болезнь Нимана-Пика
Мукополисахаридоз
Болезнь Гоше
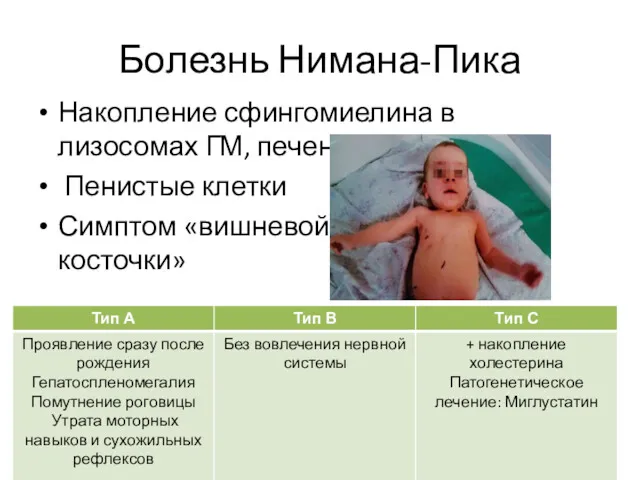
Болезнь Нимана-Пика
Накопление сфингомиелина в лизосомах ГМ, печени, РЭС.
Пенистые клетки
Симптом «вишневой
Болезнь Нимана-Пика
Накопление сфингомиелина в лизосомах ГМ, печени, РЭС.
Пенистые клетки
Симптом «вишневой
Тип С
Ювенильная форма
+ Филиппиновый тест
Тип С
Ювенильная форма
+ Филиппиновый тест
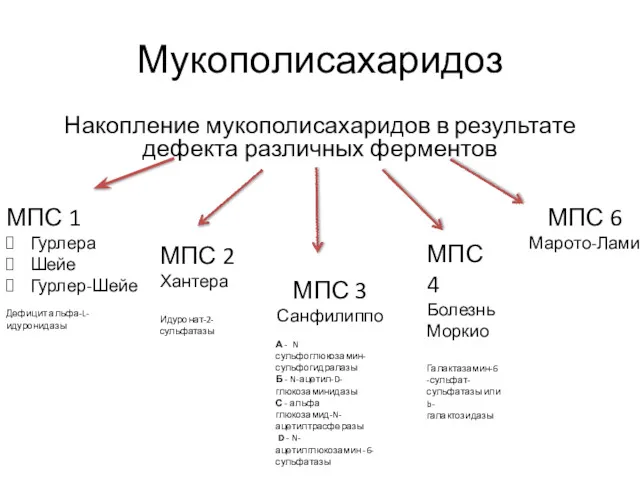
Мукополисахаридоз
Накопление мукополисахаридов в результате дефекта различных ферментов
МПС 1
Гурлера
Шейе
Гурлер-Шейе
Дефицит альфа-L-идуронидазы
МПС
Мукополисахаридоз
Накопление мукополисахаридов в результате дефекта различных ферментов
МПС 1
Гурлера
Шейе
Гурлер-Шейе
Дефицит альфа-L-идуронидазы
МПС
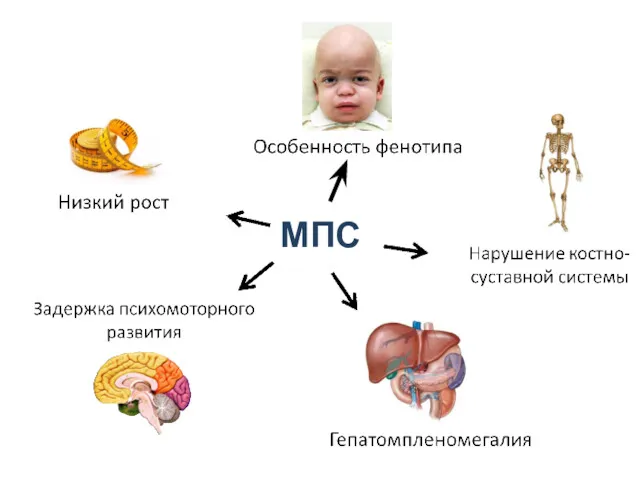
МПС
МПС
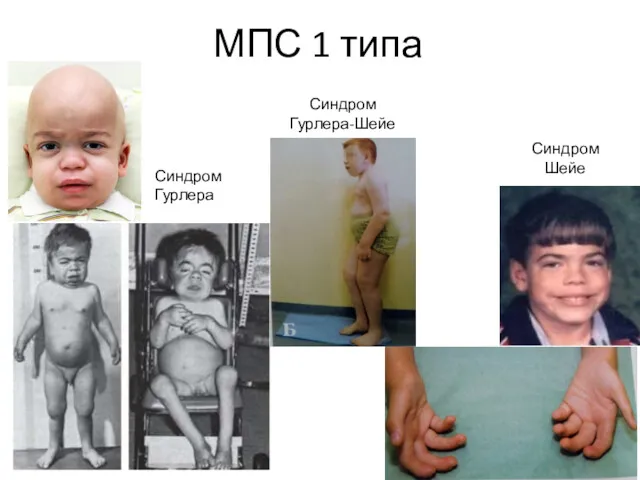
МПС 1 типа
Синдром Гурлера
Синдром Шейе
Синдром Гурлера-Шейе
МПС 1 типа
Синдром Гурлера
Синдром Шейе
Синдром Гурлера-Шейе
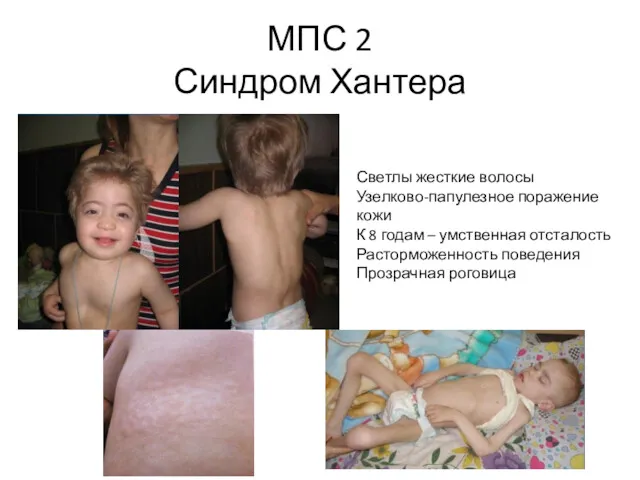
МПС 2
Синдром Хантера
Светлы жесткие волосы
Узелково-папулезное поражение кожи
К 8 годам –
МПС 2
Синдром Хантера
Светлы жесткие волосы
Узелково-папулезное поражение кожи
К 8 годам –

МПС III
Синдром Санфилиппо
К 3 годам жизни развитие прекращается
Физическое развитие соответствует
МПС III
Синдром Санфилиппо
К 3 годам жизни развитие прекращается
Физическое развитие соответствует
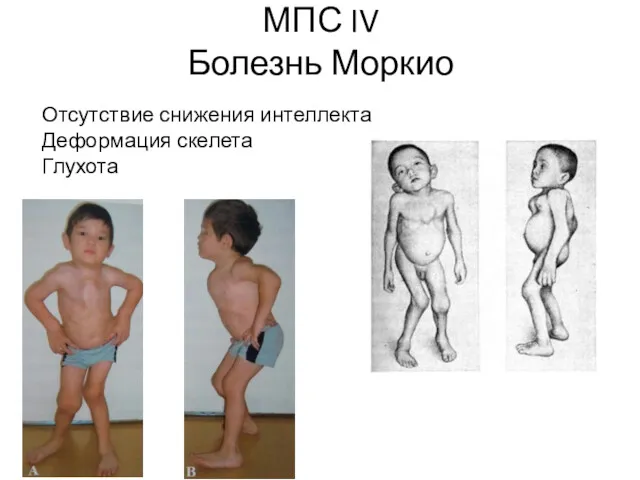
МПС IV
Болезнь Моркио
Отсутствие снижения интеллекта
Деформация скелета
Глухота
МПС IV
Болезнь Моркио
Отсутствие снижения интеллекта
Деформация скелета
Глухота
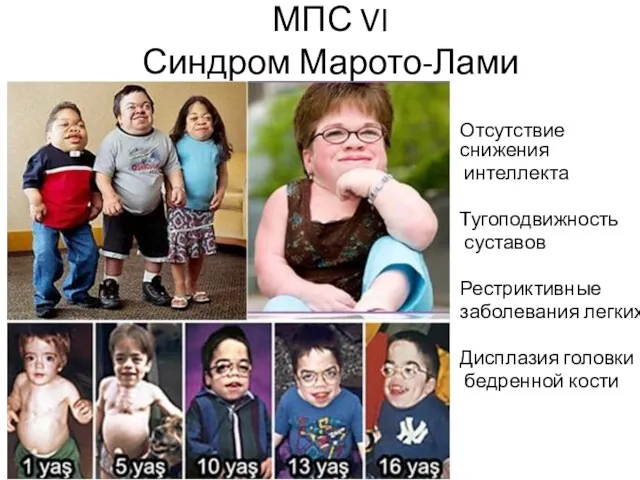
МПС VI
Синдром Марото-Лами
Отсутствие снижения
интеллекта
Тугоподвижность
суставов
Рестриктивные
заболевания легких
Дисплазия головки
бедренной кости
МПС VI
Синдром Марото-Лами
Отсутствие снижения
интеллекта
Тугоподвижность
суставов
Рестриктивные
заболевания легких
Дисплазия головки
бедренной кости
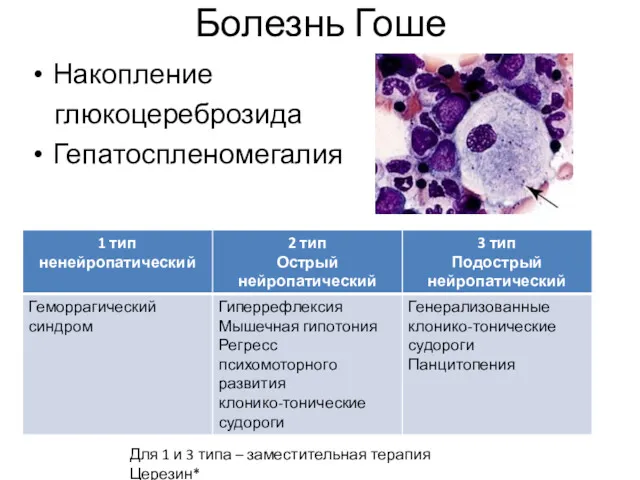
Болезнь Гоше
Накопление
глюкоцереброзида
Гепатоспленомегалия
Для 1 и 3 типа – заместительная терапия
Болезнь Гоше
Накопление
глюкоцереброзида
Гепатоспленомегалия
Для 1 и 3 типа – заместительная терапия
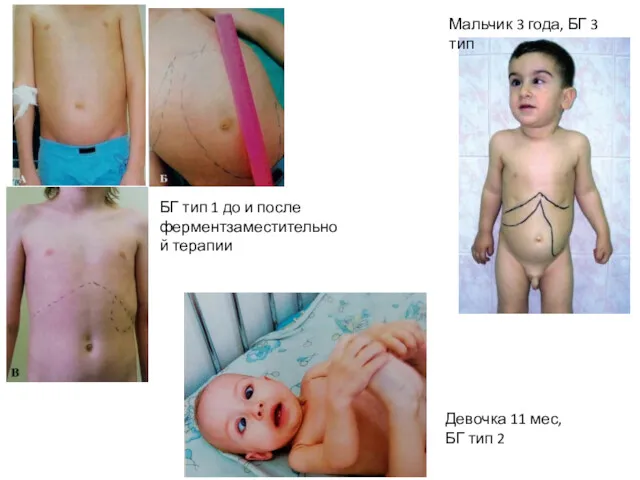
БГ тип 1 до и после ферментзаместительной терапии
Девочка 11 мес, БГ
БГ тип 1 до и после ферментзаместительной терапии
Девочка 11 мес, БГ

Митохондриальные заболевания
Феномен гетероплазии – присутствие мутантных и нормальных мтДНК в одной
Митохондриальные заболевания
Феномен гетероплазии – присутствие мутантных и нормальных мтДНК в одной

Синдром Кернса-Сейра
Наружная офтальмоплегия
Миопатия
Атаксия
Нейросенсорная глухота
Феномен «рваных красных волокон» в мышечных биоптатах
Синдром Кернса-Сейра
Наружная офтальмоплегия
Миопатия
Атаксия
Нейросенсорная глухота
Феномен «рваных красных волокон» в мышечных биоптатах
 Результаты анкетирования пациентов по ПМО с 05.03. по 14.03.2018 г. (ПО № 1)
Результаты анкетирования пациентов по ПМО с 05.03. по 14.03.2018 г. (ПО № 1) Гемофилия. Причины появления
Гемофилия. Причины появления Курація хворого з гострою серцевою недостаністю
Курація хворого з гострою серцевою недостаністю Ответственность медицинских работников по уголовному кодексу
Ответственность медицинских работников по уголовному кодексу Повышение температуры тела. Причины. Диагностика и лечение
Повышение температуры тела. Причины. Диагностика и лечение Подростковая беременность
Подростковая беременность Клиника и диагностика пролапса тазовых органов
Клиника и диагностика пролапса тазовых органов Диспансеризация взрослого населения
Диспансеризация взрослого населения Неврологический осмотр больного в коматозном состоянии
Неврологический осмотр больного в коматозном состоянии Жүйке жүйесінің инфекциялық аурулары: Менингит
Жүйке жүйесінің инфекциялық аурулары: Менингит Анализ финансовой деятельности медицинских организаций
Анализ финансовой деятельности медицинских организаций Поражения жкт при приеме НПВП
Поражения жкт при приеме НПВП Листериялар
Листериялар Вирусты гепатиттер В, Д, C
Вирусты гепатиттер В, Д, C Зеленая аптека
Зеленая аптека Диагностика и лечение неотложных состояний в онкогинекологии (кровотечение, острая кишечная непроходимость, перитонит)
Диагностика и лечение неотложных состояний в онкогинекологии (кровотечение, острая кишечная непроходимость, перитонит) Вроджені вади грудної клітки
Вроджені вади грудної клітки Преаналитический этап в лабораторной диагностике. Требования по подготовке пациента к лабораторным исследованиям
Преаналитический этап в лабораторной диагностике. Требования по подготовке пациента к лабораторным исследованиям Шизофрения ауруы
Шизофрения ауруы Вирусные гепатиты
Вирусные гепатиты Медицинская микробиология
Медицинская микробиология Микробтарға қарсы заттар. Антисептикалық және дезинфекциялаушы заттар
Микробтарға қарсы заттар. Антисептикалық және дезинфекциялаушы заттар Роль медицинской сестры в уходе за детьми с атопическим дерматитом
Роль медицинской сестры в уходе за детьми с атопическим дерматитом Недоношенные дети
Недоношенные дети Фитфуд. Решение проблем
Фитфуд. Решение проблем Гигиена труда при работе с химическими профессиональными факторами (гигиеническая токсикология)
Гигиена труда при работе с химическими профессиональными факторами (гигиеническая токсикология) Парамфистоматозы и другие широко распространенные трематодозы
Парамфистоматозы и другие широко распространенные трематодозы Грыжи. Составные элементы грыжи
Грыжи. Составные элементы грыжи