- Наследственные заболевания нервной системы

Содержание
- 2. Наследственные заболевания 1)генные: моногеннные полигенные 2) хромосомные Изменение числа аутосом синдром Дауна — трисомия по 21
- 3. Cиндром Дауна трисомия по 21 хромосоме
- 4. Синдром Патау трисомия по 13 хромосоме множественные пороки развития часто полидактилия нарушения строения половых органов глухота
- 5. Синдром Эдвардса ( трисомия по 18 хромосоме) нижняя челюсть и ротовое отверстие маленькие глазные щели узкие
- 6. Типы наследования Аутосомно-доминантный Риск рождения больного ребенка – 50%
- 7. Аутосомно-рецессивный тип наследования Риск рождения больного ребенка – 25 %
- 8. Наследование, сцепленное с Х-хромосомой Мальчики болеют Девочки - носители
- 9. Нервно-мышечные заболевания Миогенные формы (поражение непосредственно мышечной ткани) Миопатии Мышечные дистрофии Миотонии Нейрогенные формы (поражение нервной
- 10. Миогенные формы. Псевдогипертрофическая мышечная дистрофия Дюшенна Частота – 14 на 100 000 родившихся Тип наследования –
- 11. Патоморфология – перерождение мышечной ткани, замещение ее жировой и соединительной тканью.
- 12. Дебют – первые 5 лет жизни Первые признаки: слабость мышц тазового пояса дети испытывают затруднение при
- 13. Псевдогипертрофии икроножных, дельтовидных и ягодичных мышц Первый из глубоких рефлексов исчезает коленный Из-за усиливающейся слабости перестают
- 14. Псевдогипертрофии икроножных мышц
- 15. Симптом Говерса (симптом вставания «лесенкой»)
- 17. Диагностика Повышение в крови ферментов (еще в доклиническую стадию!) : КФК (креатинфосфокиназа) ЛДГ АЛТ АСТ Это
- 18. Поздняя песвдогипертрофическая форма Беккера Наследование – сцепленное с Х-хромосомой Заболевание клинически идентично миодистрофии Дюшенна, однако начало
- 19. Врожденные миопатии Патоморфологические изменения являются следствием аномалий развития и дифференцировки поперечнополосатых мышц. Болезнь центрального стержня Нитчатая
- 20. Болезнь центрального стержня Начальные признаки болезни проявляются в раннем возрасте общей мышечной гипотонией, мышечной слабостью, задержкой
- 21. Нитчатая (немалиновая) миопатия Наиболее частая из врожденных миопатий Внешние дизэмбриогенетические признаки (удлиненный лицевой череп, низко расположенные
- 22. Миотонии Феномен миотонии – непроизвольный тонический спазм мышц вслед за резким произвольным сокращением. Расслабление наступает ч/з
- 23. Атрофическая миотония Дебют более типичен на II-III десятилетии жизни. Симптом миотонического ровика легче получить на дельтовидной,
- 24. Невральные амиотрофии (наследственные мотосенсорные полинейропатии) Чаще страдает аксон II нейрона Моторные нарушения: симметричные парезы в дистальных
- 25. Невральные амиотрофии (наследственные мотосенсорные полинейропатии) Невральная амиотрофия Шарко-Мари-Тута Аут-доминантно Дебют – первое 10-летие жизни. Начальные симптомы
- 26. Невральные амиотрофии (наследственные мотосенсорные полинейропатии)
- 27. Диагностика ЭНМГ: Снижение скорости проведения возбуждения по нервному стволу. Биохимические методы исследования не выявляют особой патологии
- 28. Детские спинальные амиотрофии ТИП НАСЛЕДОВАНИЯ – АУТОСОМНО-РЕЦЕССИВНЫЙ Тип 1 – б-нь Верднига-Гоффмана (острая спинальная амиотрофия) Тип
- 29. Ритм частокола на ЭНМГ (из-за дегенерации мотонейронов спинного мозга)
- 30. Факоматозы греч. phacos —пятно группа системных заболеваний, характеризующихся поражением кожных покровов, нервной системы и часто внутренних
- 31. Классификация факоматозов бластоматозы туберозный склероз (б-нь Бурневиля-Прингла) нейрофиброматоз Реклингхаузена ангиоматозы энцефалотригеминальный ангиоматоз Штурге-Вебера атаксия-телеангиэктазия Луи-Бар цереброретинальный
- 32. Туберозный склероз (эпилойя, болезнь Бурневиля — Прингла) генетически детерминированное заболевание, характеризуется поражением нервной системы, кожи и
- 33. Этиопатогенез тип наследования - аутосомно-доминантный. спорадические случаи - 50–70 % распространенность заболевания для всех возрастов около
- 34. Кожные изменения встречаются в 100 % случаев гипопигментированные пятна ангиофибромы лица участки «шагреневой кожи» фиброзные бляшки
- 35. Гипопигментированные пятна встречаются в 90 % случаев появляются обычно в первые 2–3 года жизни число пятен
- 36. Ангиофибромы лица adenoma sebaceum встречаются в 47–90 % появляются, как правило, после 4 лет жизни
- 37. Участки «шагреневой кожи» встречаются в 21–68 % случаев появляется на втором десятилетии жизни.
- 38. Фиброзные бляшки патогномоничный кожный симптом встречаются у 25 % больных часто появляются на первом году жизни
- 39. Околоногтевая фиброма (опухоль Кёнена) встречаются у 50 % больных появляются во время или непосредственно после пубертата
- 40. Поражение ЦНС 1. Субэпендимальные узлы 2. Корковые туберсы 3. Субэпендимальная гигантоклеточная астроцитома
- 41. Поражение ЦНС
- 42. Эпилептические приступы с-м Веста (инфантильные спазмы) атипичные абсансы фокальные припадки вторично-генерализованные тонико-клонические припадки
- 43. Ретинальные гамартомы выявляются у 50 % больных дебют в первые 2 года жизни симптом «тутовой ягоды»
- 44. Гамартома диска зрительного нерва
- 45. Рабдомиома сердца Эхокардиография сердца. Рабодомиома в левом желудочке
- 46. Поражение ротовой полости Фибромы десен Дефекты эмали
- 47. Поражение почек Ангиомиолипомы (у 48% больных) Кисты (у 35% больных) Почечноклеточная карцинома (у 5% больных) Ангиомиолипома
- 48. Поражение костей
- 49. Поражение легких Лимфангиомиоматоз легкие вовлекаются в патологический процесс после 30 лет У 40% женщин с туберозным
- 50. Диагностические критерии Вторичные признаки Ангиофибромы лица или фиброзные бляшки на лбу Околоногтевые фибромы Гипопигментные пятна Участок
- 51. Нейрофиброматоз (болезнь фон Реклингхаузена) Тип наследования – аутосомно-доминантный Пятна «кофе с молоком» Нейрофибромы на коже, в
- 52. Множественные кожные нейрофибромы на коже спины
- 54. Нейрофиброматоз (болезнь фон Реклингхаузена) Пятна «кофе с молоком»
- 55. Узелки Лиша на радужке глаза
- 56. Глиома зрительного нерва Нейрофиброматоз Опухоли ЦНС
- 57. Атаксия-телеангиэктазия Луи-Барр Тип наследования – аутосомно-рецессивный Клиника: мозжечковая атаксия с детства телеангиэктазии, особенно в области конъюнктивы
- 59. Энцефалотригеминальный ангиоматоз Штурге-Вебера Тип наследования – аутосомно-доминантный Триада: 1) ангиома на коже лица (в области иннервации
- 60. Ангиома на коже лица
- 62. Скачать презентацию
 Лазеры. Лазерное излучение и его основные параметры. Лазерная медицина
Лазеры. Лазерное излучение и его основные параметры. Лазерная медицина Лептоспироз. Возбудители болезни
Лептоспироз. Возбудители болезни Понятие о сосудистых коллатералях и анастомозах. Особенности кровоснабжения органов
Понятие о сосудистых коллатералях и анастомозах. Особенности кровоснабжения органов Родовая травма новорождённых
Родовая травма новорождённых Гирудотерапия на современном этапе
Гирудотерапия на современном этапе Сестринские вмешательства при нарушении деятельности желудка
Сестринские вмешательства при нарушении деятельности желудка Артериальная гипертензия у беременных
Артериальная гипертензия у беременных ҚРда және жақын шетелде медициналық
ҚРда және жақын шетелде медициналық Катаракта и современные методы лечения
Катаракта и современные методы лечения Ұйқы бұзылысы
Ұйқы бұзылысы Презентация_Жүктілік кезіндегі гипертензивті жағдайлар..pptx
Презентация_Жүктілік кезіндегі гипертензивті жағдайлар..pptx Лечение наследственных болезней
Лечение наследственных болезней Анафилактический шок
Анафилактический шок Антропометрия, измерение основных физических показателей человека
Антропометрия, измерение основных физических показателей человека Столбняк. Клиника. Диагностика. Лечение
Столбняк. Клиника. Диагностика. Лечение Система непрерывного медицинского и фармацевтического образования
Система непрерывного медицинского и фармацевтического образования Профилактика и коррекция нарушений осанки
Профилактика и коррекция нарушений осанки Роль медицинской сестры в профилактике заражением вирусным гепатитом В в условиях стационара
Роль медицинской сестры в профилактике заражением вирусным гепатитом В в условиях стационара Мектеп жасындағы балалардың күнтізбелік тәртібі. Алты жастағы балаларды оқытуды гигиеналық ұйымдастыру қағидалары
Мектеп жасындағы балалардың күнтізбелік тәртібі. Алты жастағы балаларды оқытуды гигиеналық ұйымдастыру қағидалары Коррекция аутизма
Коррекция аутизма Sexually transmitted bacterial diseases
Sexually transmitted bacterial diseases Сон и сновидения
Сон и сновидения Иммуноферментті анализді жүргізу методикасы
Иммуноферментті анализді жүргізу методикасы Организмнің реактивтілігі
Организмнің реактивтілігі Патофизиология почек. Почечная недостаточность
Патофизиология почек. Почечная недостаточность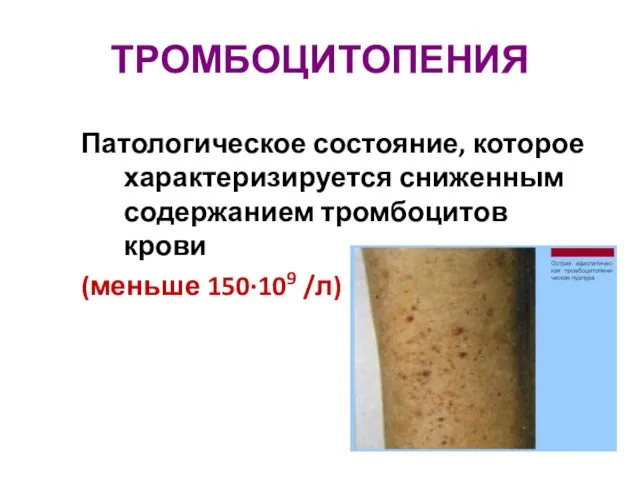 Тромбоцитопения. Классификация
Тромбоцитопения. Классификация Виды медицинской помощи
Виды медицинской помощи Проведение иммунопроилактики инфекционных заболеваний у детей
Проведение иммунопроилактики инфекционных заболеваний у детей