- Неонатальный скрининг

Содержание
- 2. Что такое неонатальный скрининг? Массовое лабораторные обследование новорожденных на некоторые врожденные заболевания
- 3. Основные принципы скрининга К общим требованиям выполнения программ скрининга новорождённых на наследственные болезни обмена, согласно документам
- 4. Этапы скрининга новорожденных 1-й этап — забор крови у новорождённых из пятки в родовспомогательных учреждениях Взятие
- 5. Этапы скрининга новорожденных 2-й этап — быстрое проведение первичного скрининга по определению соответствующих лабораторных показателей. Анализ
- 6. Этапы скрининга новорожденных 4-й этап — лечение выявленных больных, которое должны осуществлять врачи-генетики, неонатологи, педиатры и
- 7. Наследственные болезни обмена веществ: Фенилкетонурия Гипотериоз Муковисцидоз Галактоземия Гипоплазия надпочечников
- 8. Фенилкетонурия группа аутосомно-рецессивных заболеваний, обуслоленных нарушением обмена незаменимой аминокислоты фенилаланина, приводящие к поражению главным образом ЦНС.
- 9. Фенилкетонурия: патогенез Поступление фенилаланина в организм(с пищей) ДЕФЕКТНЫЙ ФЕРМЕНТ (МУТАЦИЯ ГЕНА) НЕТ ПРЕВРАЩЕНИЯ ФЕНИЛАЛАНИНА В ТИРОЗИН
- 10. Фенилкетанурия: клиника Фенилкетонурия проявляется на первом году жизни. отсутствие интереса к окружающему; повышенная раздражительность; беспокойство; срыгивания;
- 11. Лечение Диетотерапия – патогенетически обоснованный и наиболее эффективный метод лечения классической ФКУ. Диетотерапия должна быть начата
- 12. Врождённый гипотиреоз заболевание, обусловленное полным отсутствием или уменьшенной продукцией тиреоидных гормонов щитовидной железой либо снижением их
- 13. Патогенез наследственного ВГ Дефект генов на 1- ой и 22- ой хромосомах Нарушение синтеза тиреоидных гормонов
- 14. Клиническая картина вялое, неэффективное сосание плохая прибавка массы тела мышечная гипотания пониженная устойчиость к гипотермии пролонгироанная
- 15. Клиническая картина сухость и бледность кожи отечность склонность к запорам брадикардия анемия гипербилирубинемия гиперхолестеринемия
- 16. Заместительная терапия тиреоидными гормонами наиболее распространенный препарат для заместительной терапии – тироксин (L-тироксин, левотироксин), реже трийодтиронин
- 17. Гипоплазия коры надпочечников Наследственное заболевание, которое вызывается мутацией в гене, картированном на хромосоме 6р21.3. Заболевание наследуется
- 18. Патогенез Дефект гена NR0B1 Da Дефект фактора DAX1 Нарушение закладки коры надпочечников (+ яичек у мальчиков)
- 19. Клиническая картина
- 20. Муковисцидоз (кистозный фиброз) – аутосомно- рецессивное моногенное заболевание, обусловленное мутацией гена МВТР (муковисцидозный трансмембранный регулятор проводимости),
- 21. Классификация Формы МВ — смешанная (легочно-кишечная) (80% всех случаев); — с преимущественно легочными проявлениями (15%); —
- 22. Патогенез нарушение синтеза первичного продукта гена CFTR - трансмембранного регулятора проводимости ионов Мутация гена . Дисфункция
- 23. Клинические проявления Рецидивирующие или хронические респираторные симптомы, такие как кашель или одышка Повторные пневмонии Мекониальный илеус
- 24. Диагностика Неонатальная диагностика. • первое определение концентрации иммунореактивного трипсина (на первой неделе жизни); • повторное определение
- 25. Галактоземия- наследственное аутосомно-рецессивное нарушение обмена углеводов, при котором накапливается избыток галактозы и ее метаболитов, что обуславливает
- 26. Классификация Классическая - галактоземия I типа, обусловленная дефицитом фермента галактозо-1-фосфат-уридилтрансферазы (ГАЛТ). Галактоземия I I-недостаточность галактокиназы (ГАЛК).
- 27. Клиническая картина Галактоземия I тип. Манифестация на 1 -2 неделе Частые срыгивания Диарея Гепатоспленомегалия Гипербилирубинемия Гипоальбунемия
- 28. Клиническая картина Галактоземия I I тип. Единственное проявление заболевания-катаракта. Галактоземия I I I тип. Доброкачественная Тяжелая
- 29. Лечение Диетотерапия Урацил-4-карбоновая кислота Гепатопротекторы Антиоксиданты Хирургическое лечение катаракты
- 30. Скрининг в мире Ситуация в мире по количеству исследуемых нозологий:
- 31. Пилотный проект по скринингу новорожденных в Свердловской области от 2012 года: РАСШИРЕНИЕ ОБСЛЕДОВАНИЯ НОВОРОЖДЕННЫХ СВЕРДЛОВСКОЙ ОБЛАСТИ
- 32. 16 наследственных заболеваний Недостаточность очень длинных и средних цепей ацил-КoA-дегидрогеназы жирных кислот. Недостаточность митохондриального трифункционального белка.
- 34. Скачать презентацию
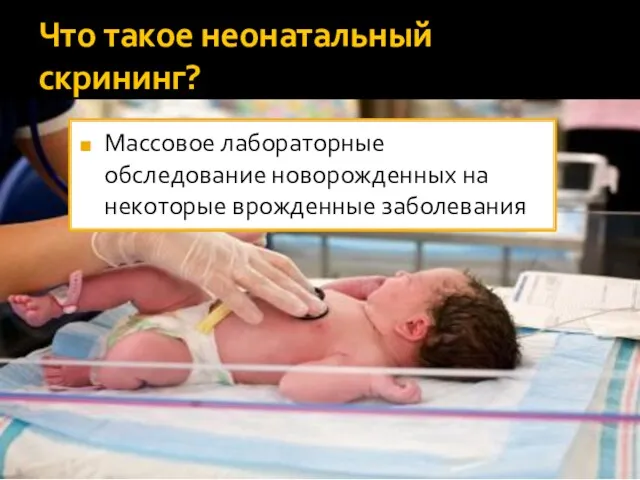
Что такое неонатальный скрининг?
Массовое лабораторные обследование новорожденных на некоторые врожденные заболевания
Что такое неонатальный скрининг?
Массовое лабораторные обследование новорожденных на некоторые врожденные заболевания

Основные принципы скрининга
К общим требованиям выполнения программ скрининга новорождённых на наследственные
Основные принципы скрининга
К общим требованиям выполнения программ скрининга новорождённых на наследственные

Этапы скрининга новорожденных
1-й этап — забор крови у новорождённых из пятки
Этапы скрининга новорожденных
1-й этап — забор крови у новорождённых из пятки

Этапы скрининга новорожденных
2-й этап — быстрое проведение первичного скрининга по определению
Этапы скрининга новорожденных
2-й этап — быстрое проведение первичного скрининга по определению

Этапы скрининга новорожденных
4-й этап — лечение выявленных больных, которое должны осуществлять
Этапы скрининга новорожденных
4-й этап — лечение выявленных больных, которое должны осуществлять

Наследственные болезни обмена веществ:
Фенилкетонурия
Гипотериоз
Муковисцидоз
Галактоземия
Гипоплазия надпочечников
Наследственные болезни обмена веществ:
Фенилкетонурия
Гипотериоз
Муковисцидоз
Галактоземия
Гипоплазия надпочечников
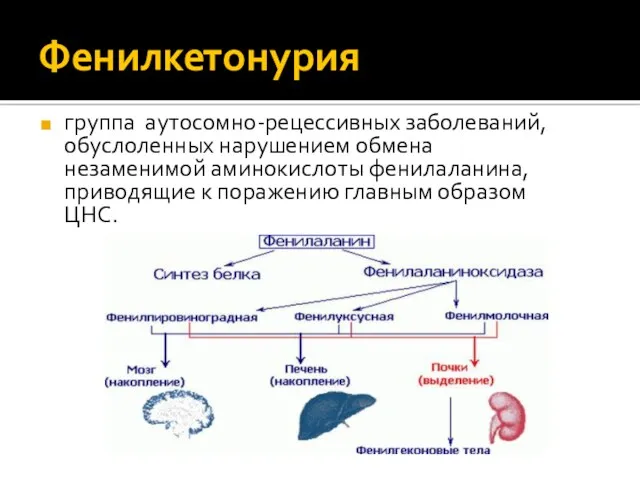
Фенилкетонурия
группа аутосомно-рецессивных заболеваний, обуслоленных нарушением обмена незаменимой аминокислоты фенилаланина, приводящие к
Фенилкетонурия
группа аутосомно-рецессивных заболеваний, обуслоленных нарушением обмена незаменимой аминокислоты фенилаланина, приводящие к

Фенилкетонурия: патогенез
Поступление фенилаланина в организм(с пищей)
ДЕФЕКТНЫЙ ФЕРМЕНТ (МУТАЦИЯ ГЕНА)
НЕТ ПРЕВРАЩЕНИЯ ФЕНИЛАЛАНИНА
Фенилкетонурия: патогенез
Поступление фенилаланина в организм(с пищей)
ДЕФЕКТНЫЙ ФЕРМЕНТ (МУТАЦИЯ ГЕНА)
НЕТ ПРЕВРАЩЕНИЯ ФЕНИЛАЛАНИНА

Фенилкетанурия: клиника
Фенилкетонурия проявляется на первом году жизни.
отсутствие интереса к окружающему;
Фенилкетанурия: клиника
Фенилкетонурия проявляется на первом году жизни.
отсутствие интереса к окружающему;

Лечение
Диетотерапия – патогенетически обоснованный и наиболее эффективный метод лечения классической ФКУ.
Лечение
Диетотерапия – патогенетически обоснованный и наиболее эффективный метод лечения классической ФКУ.

Врождённый гипотиреоз
заболевание, обусловленное полным отсутствием или уменьшенной продукцией тиреоидных гормонов щитовидной
Врождённый гипотиреоз
заболевание, обусловленное полным отсутствием или уменьшенной продукцией тиреоидных гормонов щитовидной

Патогенез наследственного ВГ
Дефект генов на 1- ой и 22- ой хромосомах
Нарушение
Патогенез наследственного ВГ
Дефект генов на 1- ой и 22- ой хромосомах
Нарушение

Клиническая картина
вялое, неэффективное сосание
плохая прибавка массы тела
мышечная гипотания
пониженная устойчиость к гипотермии
пролонгироанная
Клиническая картина
вялое, неэффективное сосание
плохая прибавка массы тела
мышечная гипотания
пониженная устойчиость к гипотермии
пролонгироанная

Клиническая картина
сухость и бледность кожи
отечность
склонность к запорам
брадикардия
анемия
гипербилирубинемия
гиперхолестеринемия
Клиническая картина
сухость и бледность кожи
отечность
склонность к запорам
брадикардия
анемия
гипербилирубинемия
гиперхолестеринемия
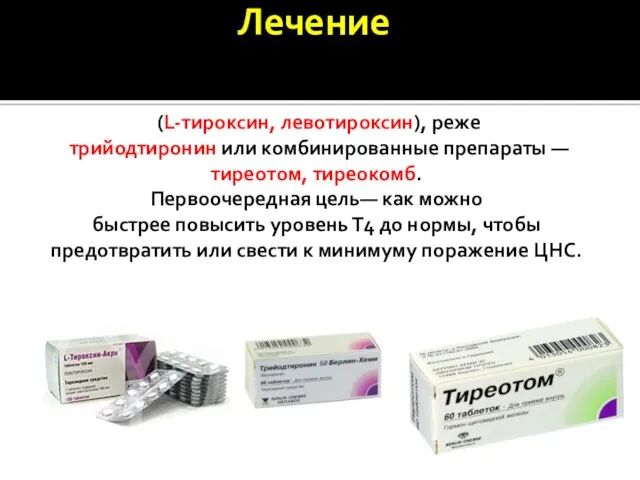
Заместительная терапия тиреоидными гормонами
наиболее распространенный препарат для заместительной терапии – тироксин
Заместительная терапия тиреоидными гормонами
наиболее распространенный препарат для заместительной терапии – тироксин

Гипоплазия коры надпочечников
Наследственное заболевание, которое вызывается мутацией в гене, картированном на
Гипоплазия коры надпочечников
Наследственное заболевание, которое вызывается мутацией в гене, картированном на
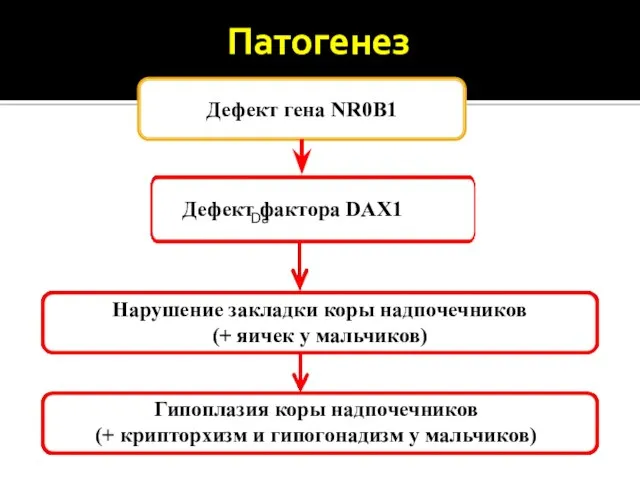
Патогенез
Дефект гена NR0B1
Da
Дефект фактора DAX1
Нарушение закладки коры надпочечников
(+ яичек у мальчиков)
Гипоплазия
Патогенез
Дефект гена NR0B1
Da
Дефект фактора DAX1
Нарушение закладки коры надпочечников
(+ яичек у мальчиков)
Гипоплазия
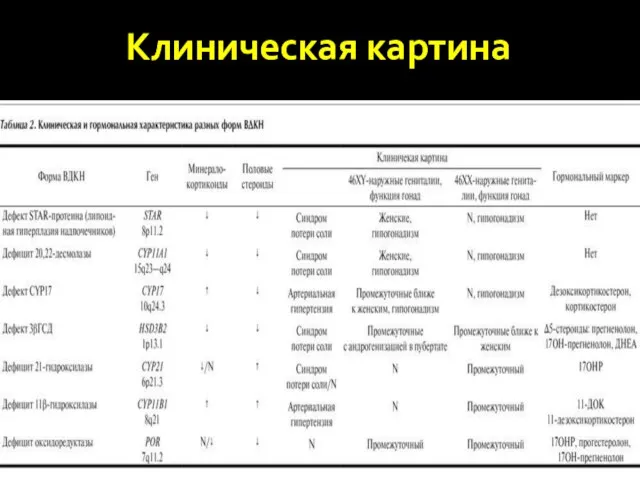
Клиническая картина
Клиническая картина
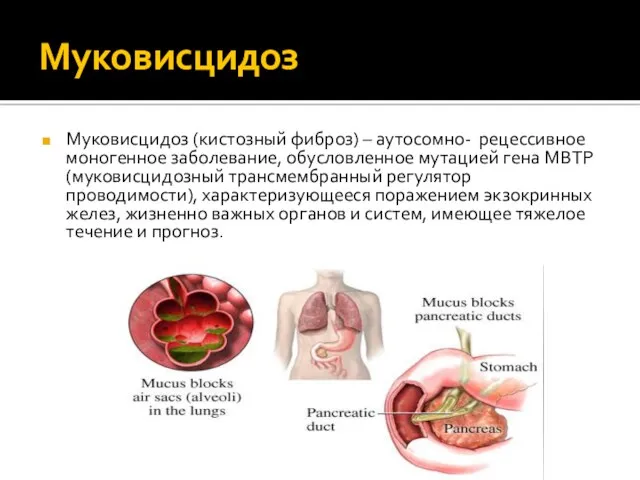
Муковисцидоз (кистозный фиброз) – аутосомно- рецессивное моногенное заболевание, обусловленное мутацией гена
Муковисцидоз (кистозный фиброз) – аутосомно- рецессивное моногенное заболевание, обусловленное мутацией гена
Классификация
Формы МВ
— смешанная (легочно-кишечная) (80% всех случаев);
— с преимущественно легочными
Классификация
Формы МВ
— смешанная (легочно-кишечная) (80% всех случаев);
— с преимущественно легочными
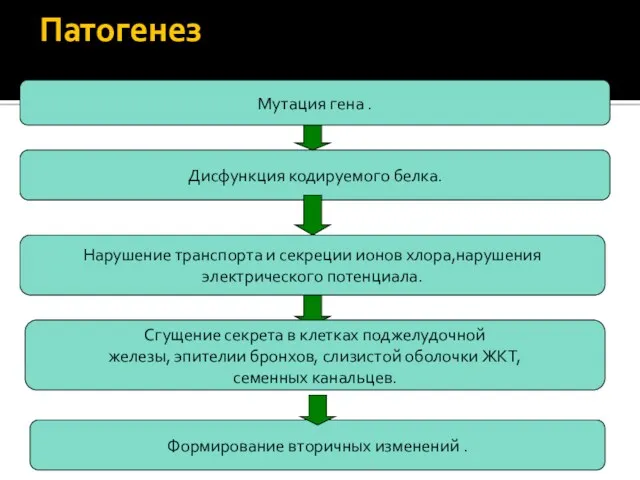
Патогенез
нарушение
синтеза первичного продукта гена CFTR
-
трансмембранного регулятора
проводимости ионов
Мутация гена
Патогенез
нарушение
синтеза первичного продукта гена CFTR
-
трансмембранного регулятора
проводимости ионов
Мутация гена

Клинические проявления
Рецидивирующие или хронические респираторные симптомы, такие как кашель или
Клинические проявления
Рецидивирующие или хронические респираторные симптомы, такие как кашель или

Диагностика
Неонатальная диагностика.
• первое определение концентрации иммунореактивного трипсина (на первой неделе жизни);
•
Диагностика
Неонатальная диагностика.
• первое определение концентрации иммунореактивного трипсина (на первой неделе жизни);
•

Галактоземия- наследственное аутосомно-рецессивное нарушение обмена углеводов, при котором накапливается избыток
Галактоземия- наследственное аутосомно-рецессивное нарушение обмена углеводов, при котором накапливается избыток

Классификация
Классическая - галактоземия I типа, обусловленная дефицитом фермента галактозо-1-фосфат-уридилтрансферазы (ГАЛТ).
Галактоземия I
Классификация
Классическая - галактоземия I типа, обусловленная дефицитом фермента галактозо-1-фосфат-уридилтрансферазы (ГАЛТ).
Галактоземия I
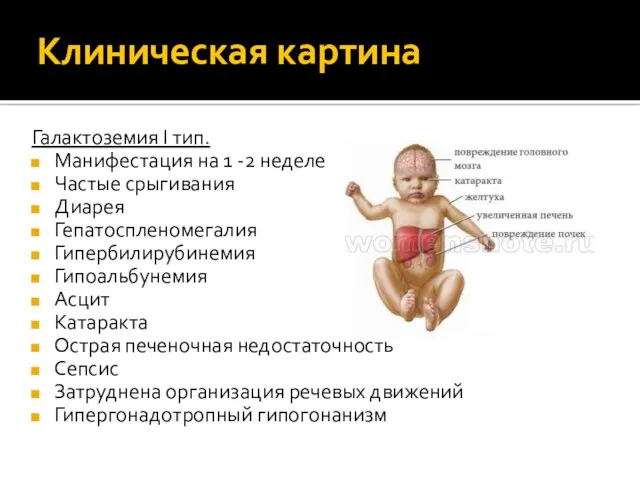
Клиническая картина
Галактоземия I тип.
Манифестация на 1 -2 неделе
Частые срыгивания
Диарея
Гепатоспленомегалия
Гипербилирубинемия
Гипоальбунемия
Асцит
Катаракта
Острая печеночная недостаточность
Сепсис
Затруднена
Клиническая картина
Галактоземия I тип.
Манифестация на 1 -2 неделе
Частые срыгивания
Диарея
Гепатоспленомегалия
Гипербилирубинемия
Гипоальбунемия
Асцит
Катаракта
Острая печеночная недостаточность
Сепсис
Затруднена

Клиническая картина
Галактоземия I I тип.
Единственное проявление заболевания-катаракта.
Галактоземия I I I тип.
Доброкачественная
Клиническая картина
Галактоземия I I тип.
Единственное проявление заболевания-катаракта.
Галактоземия I I I тип.
Доброкачественная

Лечение
Диетотерапия
Урацил-4-карбоновая кислота
Гепатопротекторы
Антиоксиданты
Хирургическое лечение катаракты
Лечение
Диетотерапия
Урацил-4-карбоновая кислота
Гепатопротекторы
Антиоксиданты
Хирургическое лечение катаракты
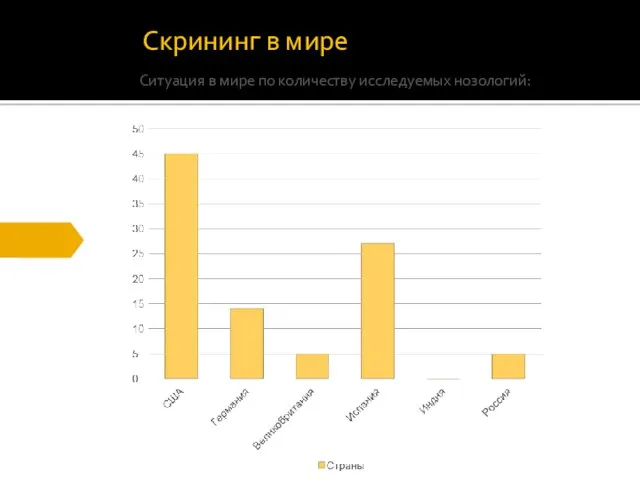
Скрининг в мире
Ситуация в мире по количеству исследуемых нозологий:
Скрининг в мире
Ситуация в мире по количеству исследуемых нозологий:

Пилотный проект по скринингу новорожденных в Свердловской области от 2012 года:
РАСШИРЕНИЕ
Пилотный проект по скринингу новорожденных в Свердловской области от 2012 года:
РАСШИРЕНИЕ

16 наследственных заболеваний
Недостаточность очень длинных и средних цепей ацил-КoA-дегидрогеназы жирных кислот.
Недостаточность
16 наследственных заболеваний
Недостаточность очень длинных и средних цепей ацил-КoA-дегидрогеназы жирных кислот.
Недостаточность
 Иерсиниозы. Этиология. Симптомы и течение
Иерсиниозы. Этиология. Симптомы и течение Нарушение кислотно-щелочного равновесия
Нарушение кислотно-щелочного равновесия Паранеопластические синдромы
Паранеопластические синдромы Адам ағзасының қалыпты микрофлорасы. Дисбактериоз
Адам ағзасының қалыпты микрофлорасы. Дисбактериоз Профилактика ОРВИ и гриппа
Профилактика ОРВИ и гриппа Медицинские капсулы, их виды и характеристика. Технологические схемы производства. Оценка качества
Медицинские капсулы, их виды и характеристика. Технологические схемы производства. Оценка качества Общая гигиена. Климат. (Лекция 12-13)
Общая гигиена. Климат. (Лекция 12-13) Основы лечебной физической культуры
Основы лечебной физической культуры Недоношенный новорожденный
Недоношенный новорожденный Анатомо-физиологические особенности органов сердечно-сосудистой системы у здорового подростка
Анатомо-физиологические особенности органов сердечно-сосудистой системы у здорового подростка Медицинская статистика. Введение. (Лекция 1)
Медицинская статистика. Введение. (Лекция 1) Частичное вторичное отсутствие зубов на нижней челюсти
Частичное вторичное отсутствие зубов на нижней челюсти Прионные болезни человека
Прионные болезни человека Лечение больных с иммунодефицитами
Лечение больных с иммунодефицитами Физиология беременности. Перинатальная охрана плода. Методы обследования в акушерстве
Физиология беременности. Перинатальная охрана плода. Методы обследования в акушерстве Проблемы ухода за пациентом с переломами при длительной иммобилизации
Проблемы ухода за пациентом с переломами при длительной иммобилизации Дыхательная недостаточность
Дыхательная недостаточность Помощь при рвоте, кормление тяжело больного пациента
Помощь при рвоте, кормление тяжело больного пациента Несеп–жыныс жүйесінің патофизиологиясы
Несеп–жыныс жүйесінің патофизиологиясы Санитарно-эпидемиологический режим в лечебных учреждениях
Санитарно-эпидемиологический режим в лечебных учреждениях Правила наложения повязок
Правила наложения повязок Дыхательная гимнастика по методике А.Н. Стрельниковой
Дыхательная гимнастика по методике А.Н. Стрельниковой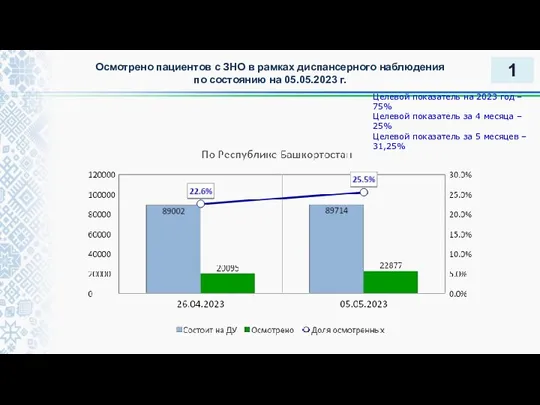 Осмотрено пациентов с ЗНО в рамках диспансерного наблюдения
Осмотрено пациентов с ЗНО в рамках диспансерного наблюдения Саңырауқұлақты аурулар. Балаларда ауыз қуысы кілегей қабатының жедел және созылмалы кандидозы. Клиникасы, емі
Саңырауқұлақты аурулар. Балаларда ауыз қуысы кілегей қабатының жедел және созылмалы кандидозы. Клиникасы, емі Санаторно-курортное обслуживание населения в СССР
Санаторно-курортное обслуживание населения в СССР Острый и хронический холецистит, желчекаменная болезнь и их осложнения
Острый и хронический холецистит, желчекаменная болезнь и их осложнения Развитие беременности и изменения, происходящие в организме женщины во время беременности. Лекция № 1
Развитие беременности и изменения, происходящие в организме женщины во время беременности. Лекция № 1 Диагностика анемий
Диагностика анемий