- Нейродегенеративные заболевания

Содержание
- 2. Распространенность в США
- 3. Болезнь Альцгеймера Открыл Алоис Альцгеймер в 1907 году Болеет 10% людей в возрасте старше 65 лет
- 4. Эпидемиология 1. Генетический компонент Наследственные формы – около 10% (аутосомно-доминантный тип) У людей с синдромом Дауна
- 5. Болезнь Альцгеймера Основные участники: белок-предшественник β-амилоида (β-APP) Tau белок Белки, участвующие в процессинге β-APP (α, β,
- 6. Tau белок Функции: стабилизирует микротрубочки в нейронах Необходим для аксонального транспорта везикул Необходим для формирования аксонов
- 7. Повреждения нейронов В нейронах накапливается большое количество «клубков», они заполняют цитоплазму, Ядро смещается Такие нейроны могут
- 8. Функции βAPP Белок-предшественник пептида бета-амилоида В норме участвует в синаптическом транспорте Нейропротектор В мутантном АРР происходит
- 9. Расщепление АРР в норме и патологии В норме происходит расщепление α-секретазой => α-APP При Альцгеймере происходит
- 10. β-APP
- 11. Агрегаты при болезни Альцгеймера
- 12. Нейротоксичность бета-амилоида Агрегация пептида бета-амилоида приводит к: Нарушениям кальциевого гомеостаза Повреждение митохондрий – окислительный стресс Воспалительный
- 13. Что происходит в мозге? Нейродегенерация начинается примерно лет за 20-30 до появления симптомов Сначала появляются клубки
- 14. Как выглядит пораженный мозг?
- 15. Этапы развития болезни 1. Первые признаки незначительны: Легкая потеря кратковременной памяти Небольшие изменения в личности (обычно
- 16. Позитронная эмиссионная томография (PET) Позволяет визуализировать активность мозга в момент совершения им когнитивных функций – запоминания-вспоминания,
- 17. Лечение амилоидных болезней В настоящий момент неизлечимы Для некоторых амилоидозов применяют трансплантацию органов (печень) Иммунологический подход
- 18. Подходы к разработке лечения А – стабилизация нативной структуры B – ингибирование протеаз, генерирующих амилоидные пептиды
- 19. Подходы к лечению Анти-ацетилхолинэстеразы замедляют развитие Хелаторы железа и меди – уменьшают окислительный стресс Ингибиторы β-
- 20. Хорея Гентингтона
- 21. Хорея Гентингтона Болезнь Гентингтона (синдром Гентингтона, хорея Гентингтона или Хантингтона) — генетическое заболевание нервной системы, характеризующееся
- 22. История 1872 George Huntington статья «On Chorea» 1932 P.R.Vessie, Davenport – статистика. Мутантный ген завезли в
- 23. Характеристика 4-10 случаев на 100,000 человек аутосомно-доминантное наследование спонтанные (новые) мутации редки (не более 10 %);
- 24. Ген Хантингтон htt или IT15 Хромосома 4, позиция 16.3 Ген располагается между 3,076,407 и 3,245,686 пн.
- 25. Первые 600 оснований гена Хантингтон (всего 180 kb) : 1 TTGCTGTGTG AGGCAGAACC TGCGGGGGCA GGGGCGGGCT GGTTCCCTGG CCAGCCATTG
- 26. Возможный механизм увеличения числа повторов – мутации экспансии Вследствие скользящего нарушения спаривания (slipping mispairing) родительской и
- 27. Белок СAG триплет кодирует аминокислоту глутамин (Gln, Q) Серия из СAG триплетов кодирует цепочку из АК
- 28. Основные свойства белка 3144 аминокислоты, изоэлектрическая точка 5.81 молекулярный вес 347 859 Da Локализация: в основном
- 29. Структурные особенности белка Нормальный белок содержит около 10 HEAT доменов (Huntington, Elongation Factor 3, PR65/A, TOR).
- 30. Кристаллографическая структура N-конца Htt. Полиглутаминовая область расположена вблизи N-конца белка.
- 31. NH2 aa3144 PolyQ P aa1 Cleavage by caspase 2, 3, 6 calpain N-terminal fragment C-terminal fragment
- 32. Взаимодействие Htt с более чем 100 белками, по крайней мере с 19 белками напрямую. Из которых
- 33. Белок Hap-1 (Huntingtin-associated protein 1) связывается с mHtt пропорционально количеству глутамина в глутамин повторяющейся области. Это
- 34. Белок Hip-1 (Huntingtin Interacting Protein) Предполагается, что высокий уровень концентрации свободной (не связанной с Htt) формы
- 35. Взаимодействие с нейротрофическим фактором мозга BDNF (Brain-derived neurotrophic factor) необходим для развития и поддержания нейронов. Htt
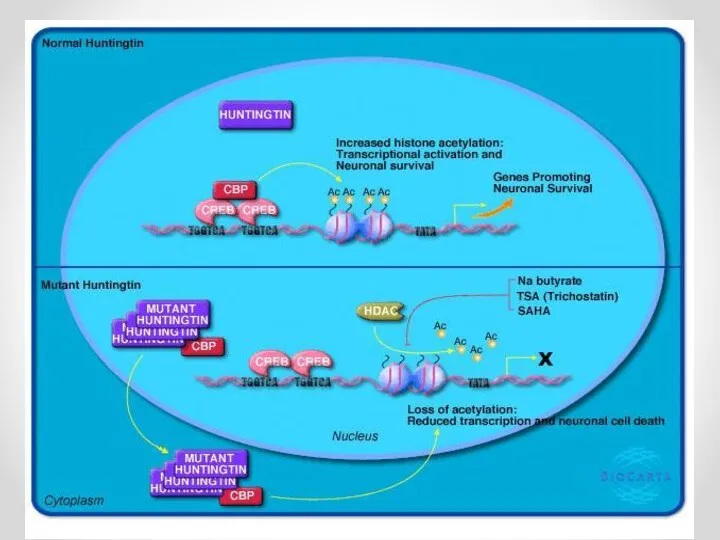
- 36. Взаимодействие с транскрипционными факторами p53,CREB mHtt связывается с p53 («страж генома» - вызывает апоптоз при нарушениях),
- 37. Взаимодействие с шаперонами и каспазами Взаимодействие mHtt с шаперонами приводит к нарушению процесса сворачивания белков. Взаимодействие
- 38. Кальциевый гомеостаз У больных нарушен кальциевый гомеостаз клеток. Увеличение концентрации ионов Ca приводит к разрушительным последствиям.
- 39. Взаимодействие с транскрипционными факторами p53,CREB mHtt связывается с p53 («страж генома» - вызывает апоптоз при нарушениях),
- 41. Взаимодействие mHtt с рецептором инозитол – 1,4,5 – трифосфата InsP3R Вызывает высвобождение ионов кальция из внутриклеточных
- 42. Potential mechanism of cell death in Huntington’s disease
- 45. Функции полосатого тела (corpus striatum) регулирует мышечный тонус, уменьшая его; участвует в регуляции работы внутренних органов;
- 46. Последствия разрушения полосатого тела 1. гипертонус скелетных мышц. 2. нарушение сложных двигательных реакций и пищедобывающего поведения.
- 47. Тип пораженных клеток Полосатое тело на 96 процентов состоит из срединных шипиковых нейронов (medium spiny neurons)
- 48. HD Normal Уменьшение мозга на 30% при Хорее Гентингтона
- 49. Физические симптомы Постепенное начало в возрасте 35-44 года; Начальная стадия заболевания: - резкие, хаотичные, бесконтрольные и
- 50. Психические симптомы Когнитивная сфера: - поражаются способности управления деятельностью и поведением: умение планировать, следовать правилам; -
- 51. Medication Antipsychotics (hallucinations, delusions, violent outbursts): haloperidol, chlorpromazine, olanzapine (contraindicated if patient has dystonia) Antidepressants (depression,
- 52. Potential therapeutic strategies in HD
- 53. Болезнь Паркинсона
- 54. Болезнь Паркинсона Вторая по распространенности среди нейродегенеративных болезней α-синуклеин формирует амилоидные фибриллы Накапливается в тельцах Lewy
- 55. Болезнь Паркинсона - хроническое нейродегенеративное заболевание, связанное с нарушением деятельности базальных ганглиев головного мозга Впервые описано
- 56. Распространенность В России насчитывается до 350000 больных болезнью Паркинсона. В США болезнью Паркинсона страдает около 500000
- 57. Эпидемиология болезни Паркинсона В мире в целом насчитывается около 6 миллионов пациентов с болезнью Паркинсона В
- 58. Причины болезни Паркинсона Старение Тот факт, что некоторые проявления болезни Паркинсона возникают и при нормальном старении,
- 59. Главные клинические проявления при болезни Паркинсона Классическая тетрада моторных признаков гипокинезия (брадикинезия и олигокинезия); мышечная ригидность
- 60. Базальные ганглии Базальные ганглии объединяют структуры: хвостатое ядро, скорлупу (вместе - полосатое тело), бледный шар и
- 61. Синтез дофамина и адреналина 1 ДОФА декарбоксилаза 2 3,4-диоксифенилаланин
- 62. При болезни Паркинсона тельца Леви в первую очередь наблюдаются в области черной субстанции - где они
- 63. Cчитается, что характерные для БП клинические признаки проявляются при гибели приблизительно 60% дофаминергических нейронов компактной части
- 64. Дофамин производится в дофаминергических нейронах, которых в мозге около 7 тысяч. Дофаминергические нейроны (как и многие
- 65. Первый ген болезни Паркинсона – ген SNCA (Polymeropoulos et al, 1967) Только мутация А53Т найдена более
- 66. Альфа-синуклеин
- 68. Тельца Леви При болезни Паркинсона в цитоплазме дофаминергических нейронов образуются тельца Леви, которые описываются как агрегаты,
- 69. Идентифицирован в 1997 г. (Tohru, et. al., Nature) 12 экзонов, 465 AA E3 убиквитин лигаза Непосредственной
- 70. Мутации в белке-паркине Показано, что белок паркин является важнейшим звеном системы клеточной защиты и, в частности,
- 71. Белок с мисфолдингом
- 72. Гены, дефекты в которых приводят к БП Гены транспорта и метаболизма дофамина • моноаминоксидазы А и
- 73. Связь между старением и болезнью Паркинсона Исследуя мутации в митохондриальной ДНК (мтДНК) нейронов мозга, две независимых
- 74. Лечение болезни Паркинсона Средства с антиоксидантным эффектом (альфа-токоферол, тиоктовая кислота, десфероксамин, ингибиторы моноаминоксидазы (МАО) типа В;
- 76. РАННЯЯ ДИАГНОСТИКА В принципе есть идеальный метод диагностики дисфункции ДА-нейронов - ПЭТ или ОФЭКТ сканирование с
- 77. Массовый скрининг: быстро - просто - относительно дешево Нарушение обоняния Обстипация (запоры) Нарушения сна (парадоксальный сон
- 78. Роль диеты в профилактике нейродегенеративных заболеваний Согласно результатам недавнего исследования, регулярное употребление рыбы, фруктов и овощей
- 80. Скачать презентацию

Распространенность в США
Распространенность в США
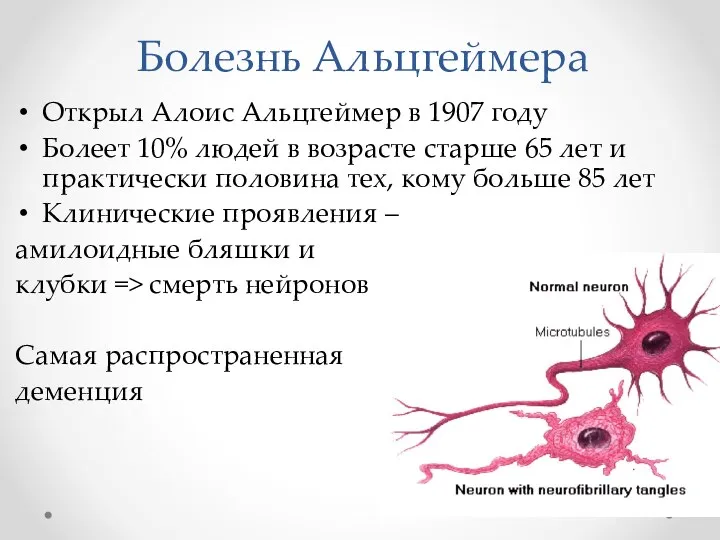
Болезнь Альцгеймера
Открыл Алоис Альцгеймер в 1907 году
Болеет 10% людей в возрасте
Болезнь Альцгеймера
Открыл Алоис Альцгеймер в 1907 году
Болеет 10% людей в возрасте

Эпидемиология
1. Генетический компонент
Наследственные формы – около 10% (аутосомно-доминантный тип)
У людей с
Эпидемиология
1. Генетический компонент
Наследственные формы – около 10% (аутосомно-доминантный тип)
У людей с

Болезнь Альцгеймера
Основные участники:
белок-предшественник β-амилоида (β-APP)
Tau белок
Белки, участвующие в процессинге β-APP
Болезнь Альцгеймера
Основные участники:
белок-предшественник β-амилоида (β-APP)
Tau белок
Белки, участвующие в процессинге β-APP

Tau белок
Функции:
стабилизирует микротрубочки в нейронах
Необходим для аксонального транспорта везикул
Необходим для формирования
Tau белок
Функции:
стабилизирует микротрубочки в нейронах
Необходим для аксонального транспорта везикул
Необходим для формирования
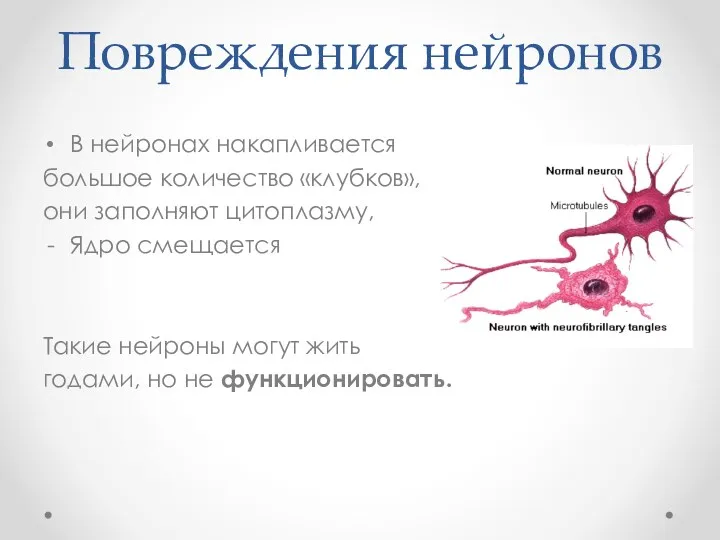
Повреждения нейронов
В нейронах накапливается
большое количество «клубков»,
они заполняют цитоплазму,
Ядро смещается
Такие
Повреждения нейронов
В нейронах накапливается
большое количество «клубков»,
они заполняют цитоплазму,
Ядро смещается
Такие

Функции βAPP
Белок-предшественник пептида бета-амилоида
В норме участвует в синаптическом транспорте
Нейропротектор
В мутантном АРР
Функции βAPP
Белок-предшественник пептида бета-амилоида
В норме участвует в синаптическом транспорте
Нейропротектор
В мутантном АРР

Расщепление АРР в норме и патологии
В норме происходит расщепление α-секретазой =>
Расщепление АРР в норме и патологии
В норме происходит расщепление α-секретазой =>
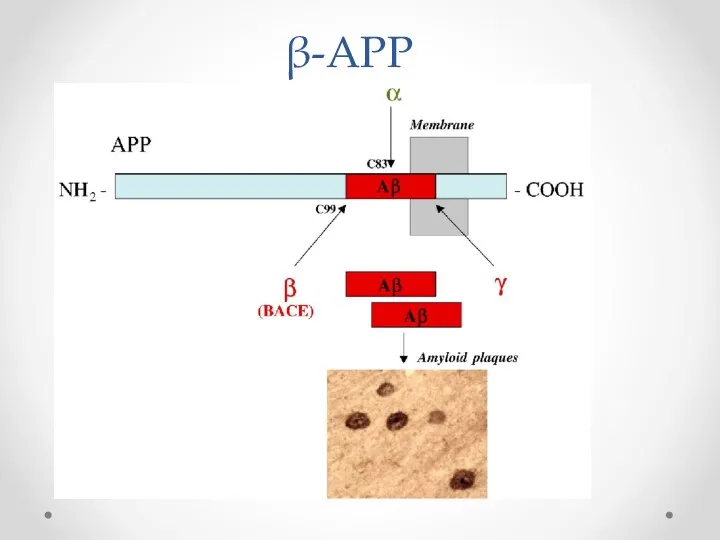
β-APP
β-APP
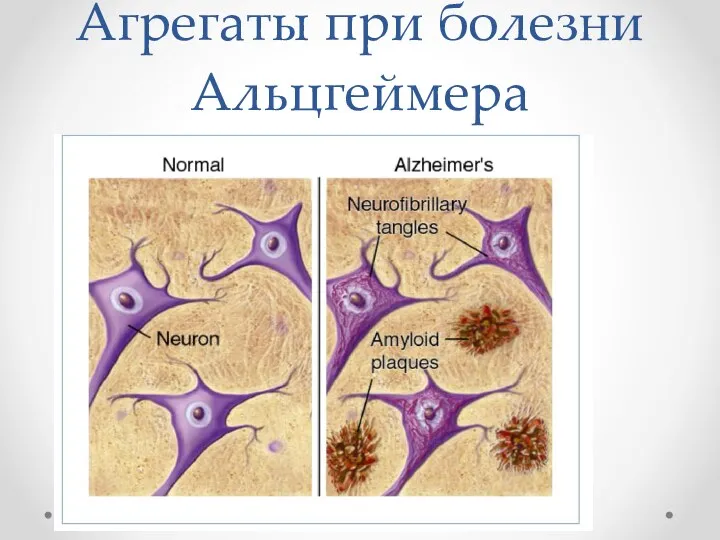
Агрегаты при болезни Альцгеймера
Агрегаты при болезни Альцгеймера

Нейротоксичность бета-амилоида
Агрегация пептида бета-амилоида приводит к:
Нарушениям кальциевого гомеостаза
Повреждение митохондрий –
Нейротоксичность бета-амилоида
Агрегация пептида бета-амилоида приводит к:
Нарушениям кальциевого гомеостаза
Повреждение митохондрий –
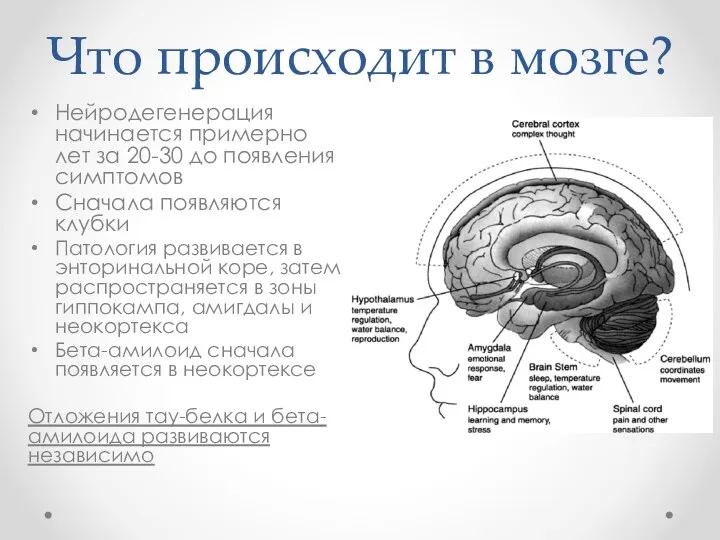
Что происходит в мозге?
Нейродегенерация начинается примерно лет за 20-30 до появления
Что происходит в мозге?
Нейродегенерация начинается примерно лет за 20-30 до появления
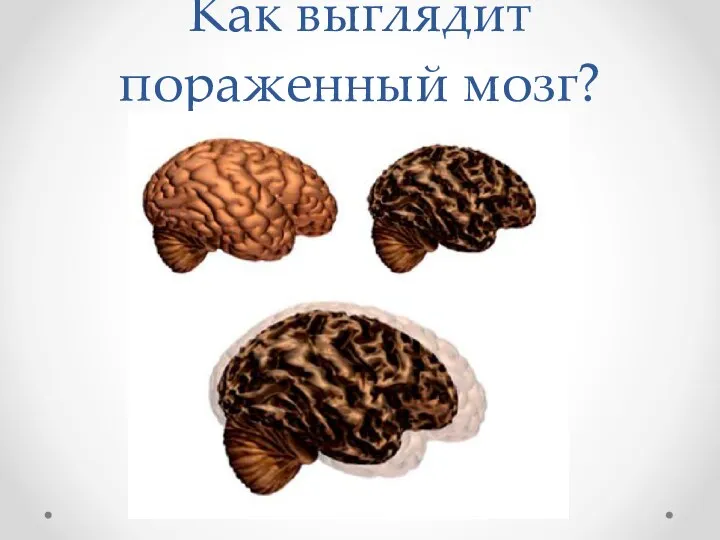
Как выглядит пораженный мозг?
Как выглядит пораженный мозг?

Этапы развития болезни
1. Первые признаки незначительны:
Легкая потеря кратковременной памяти
Небольшие изменения в
Этапы развития болезни
1. Первые признаки незначительны:
Легкая потеря кратковременной памяти
Небольшие изменения в
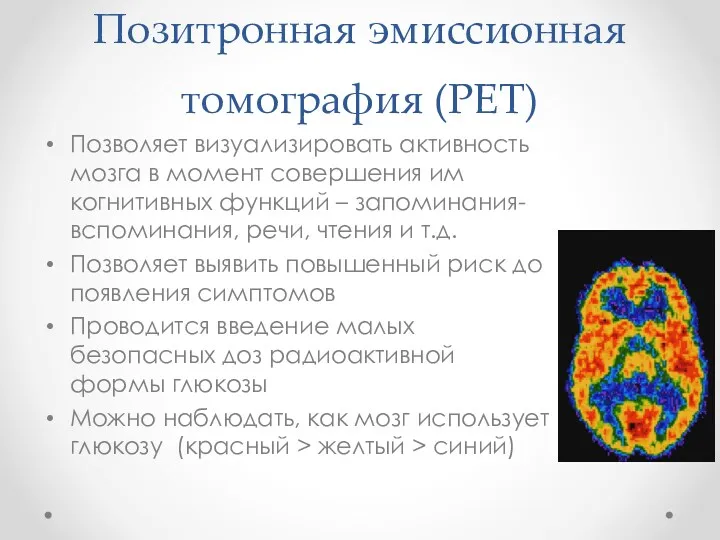
Позитронная эмиссионная томография (PET)
Позволяет визуализировать активность мозга в момент совершения им
Позитронная эмиссионная томография (PET)
Позволяет визуализировать активность мозга в момент совершения им

Лечение амилоидных болезней
В настоящий момент неизлечимы
Для некоторых амилоидозов применяют трансплантацию органов
Лечение амилоидных болезней
В настоящий момент неизлечимы
Для некоторых амилоидозов применяют трансплантацию органов
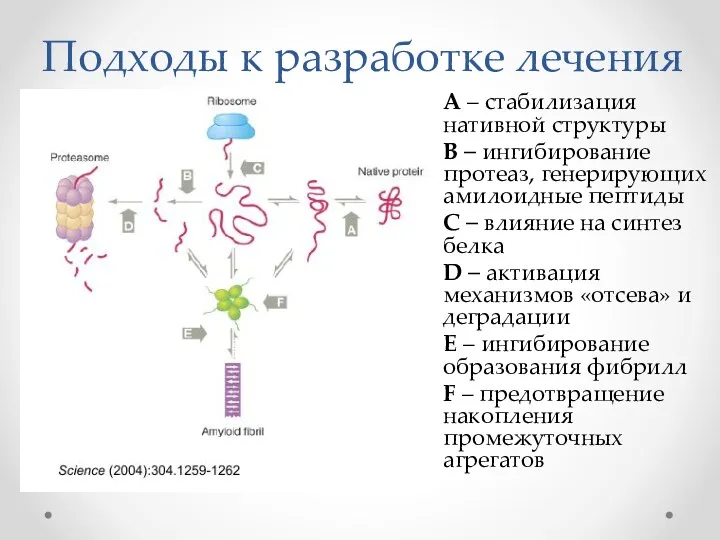
Подходы к разработке лечения
А – стабилизация нативной структуры
B – ингибирование протеаз,
Подходы к разработке лечения
А – стабилизация нативной структуры
B – ингибирование протеаз,

Подходы к лечению
Анти-ацетилхолинэстеразы замедляют развитие
Хелаторы железа и меди – уменьшают окислительный
Подходы к лечению
Анти-ацетилхолинэстеразы замедляют развитие
Хелаторы железа и меди – уменьшают окислительный

Хорея Гентингтона
Хорея Гентингтона

Хорея Гентингтона
Болезнь Гентингтона (синдром Гентингтона, хорея Гентингтона или Хантингтона) — генетическое
Хорея Гентингтона
Болезнь Гентингтона (синдром Гентингтона, хорея Гентингтона или Хантингтона) — генетическое

История
1872 George Huntington статья «On Chorea»
1932 P.R.Vessie, Davenport – статистика.
Мутантный
История
1872 George Huntington статья «On Chorea»
1932 P.R.Vessie, Davenport – статистика.
Мутантный

Характеристика
4-10 случаев на 100,000 человек
аутосомно-доминантное наследование
спонтанные (новые) мутации редки (не
Характеристика
4-10 случаев на 100,000 человек
аутосомно-доминантное наследование
спонтанные (новые) мутации редки (не

Ген Хантингтон htt или IT15
Хромосома 4, позиция 16.3
Ген располагается между 3,076,407
Ген Хантингтон htt или IT15
Хромосома 4, позиция 16.3
Ген располагается между 3,076,407

Первые 600 оснований гена Хантингтон (всего 180 kb) :
1 TTGCTGTGTG AGGCAGAACC
Первые 600 оснований гена Хантингтон (всего 180 kb) :
1 TTGCTGTGTG AGGCAGAACC

Возможный механизм увеличения числа повторов – мутации экспансии
Вследствие скользящего нарушения спаривания
Возможный механизм увеличения числа повторов – мутации экспансии
Вследствие скользящего нарушения спаривания

Белок
СAG триплет кодирует аминокислоту глутамин (Gln, Q)
Серия из СAG триплетов кодирует
Белок
СAG триплет кодирует аминокислоту глутамин (Gln, Q)
Серия из СAG триплетов кодирует

Основные свойства белка
3144 аминокислоты, изоэлектрическая точка 5.81
молекулярный вес 347 859 Da
Локализация:
Основные свойства белка
3144 аминокислоты, изоэлектрическая точка 5.81
молекулярный вес 347 859 Da
Локализация:

Структурные особенности белка
Нормальный белок содержит около 10 HEAT доменов (Huntington, Elongation
Структурные особенности белка
Нормальный белок содержит около 10 HEAT доменов (Huntington, Elongation

Кристаллографическая структура
N-конца Htt.
Полиглутаминовая область расположена вблизи N-конца белка.
Кристаллографическая структура
N-конца Htt.
Полиглутаминовая область расположена вблизи N-конца белка.
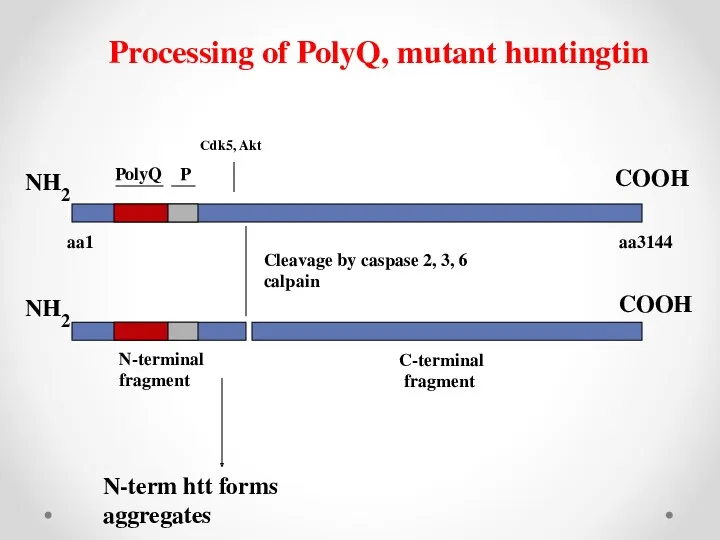
NH2
aa3144
PolyQ
P
aa1
Cleavage by caspase 2, 3, 6
calpain
N-terminal
fragment
C-terminal
fragment
COOH
COOH
NH2
Processing of PolyQ, mutant
NH2
aa3144
PolyQ
P
aa1
Cleavage by caspase 2, 3, 6
calpain
N-terminal
fragment
C-terminal
fragment
COOH
COOH
NH2
Processing of PolyQ, mutant

Взаимодействие Htt
с более чем 100 белками, по крайней мере с 19
Взаимодействие Htt
с более чем 100 белками, по крайней мере с 19

Белок Hap-1
(Huntingtin-associated protein 1)
связывается с mHtt пропорционально количеству глутамина в
Белок Hap-1
(Huntingtin-associated protein 1)
связывается с mHtt пропорционально количеству глутамина в

Белок Hip-1
(Huntingtin Interacting Protein)
Предполагается, что высокий уровень концентрации свободной (не
Белок Hip-1
(Huntingtin Interacting Protein)
Предполагается, что высокий уровень концентрации свободной (не

Взаимодействие с нейротрофическим фактором мозга
BDNF (Brain-derived neurotrophic factor) необходим для развития
Взаимодействие с нейротрофическим фактором мозга
BDNF (Brain-derived neurotrophic factor) необходим для развития

Взаимодействие с транскрипционными факторами p53,CREB
mHtt связывается с p53 («страж генома» -
Взаимодействие с транскрипционными факторами p53,CREB
mHtt связывается с p53 («страж генома» -

Взаимодействие с шаперонами и каспазами
Взаимодействие mHtt с шаперонами приводит к нарушению
Взаимодействие с шаперонами и каспазами
Взаимодействие mHtt с шаперонами приводит к нарушению

Кальциевый гомеостаз
У больных нарушен кальциевый гомеостаз клеток.
Увеличение концентрации ионов Ca
Кальциевый гомеостаз
У больных нарушен кальциевый гомеостаз клеток.
Увеличение концентрации ионов Ca

Взаимодействие с транскрипционными факторами p53,CREB
mHtt связывается с p53 («страж генома» -
Взаимодействие с транскрипционными факторами p53,CREB
mHtt связывается с p53 («страж генома» -

Взаимодействие mHtt с рецептором инозитол – 1,4,5 – трифосфата InsP3R
Вызывает
Взаимодействие mHtt с рецептором инозитол – 1,4,5 – трифосфата InsP3R
Вызывает
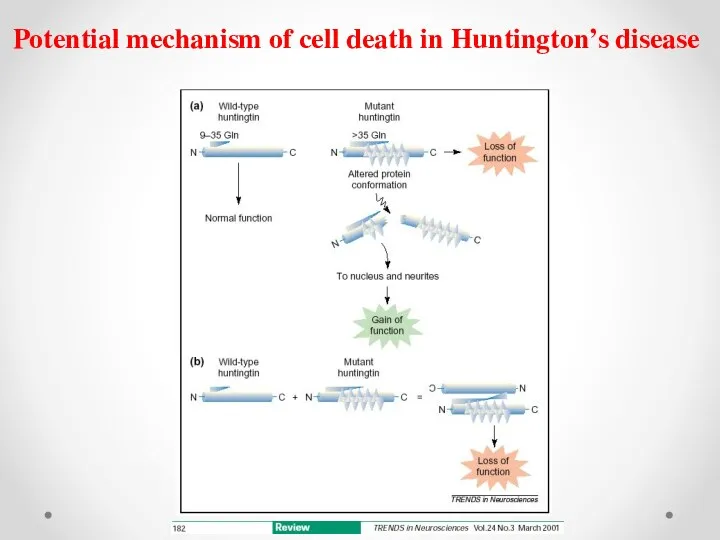
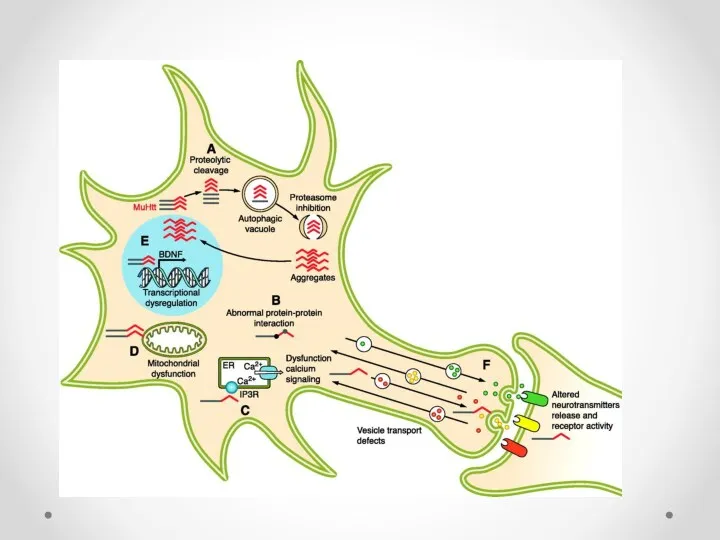
Potential mechanism of cell death in Huntington’s disease
Potential mechanism of cell death in Huntington’s disease
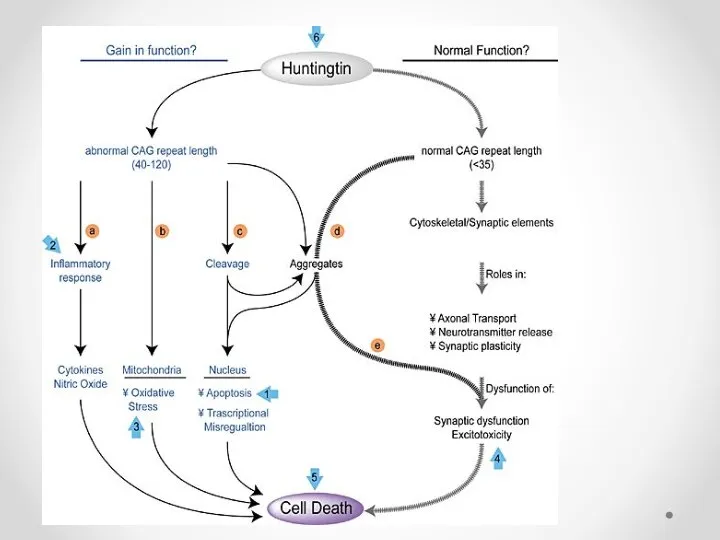

Функции полосатого тела (corpus striatum)
регулирует мышечный тонус, уменьшая его;
участвует в
Функции полосатого тела (corpus striatum)
регулирует мышечный тонус, уменьшая его;
участвует в
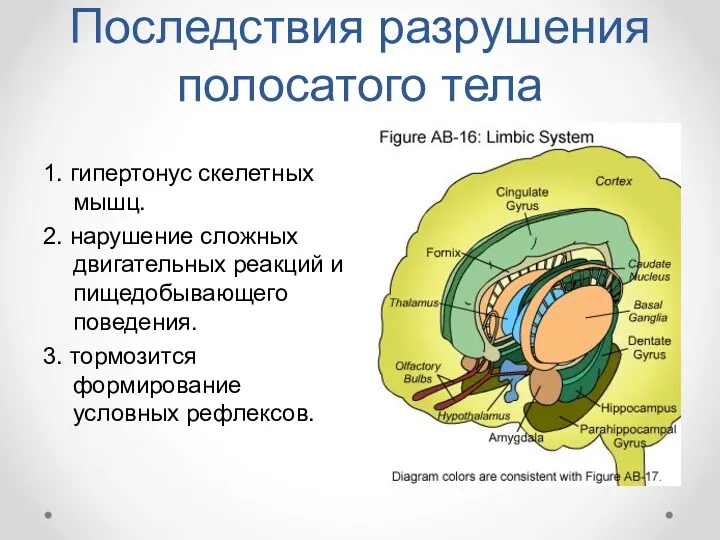
Последствия разрушения полосатого тела
1. гипертонус скелетных мышц.
2. нарушение сложных двигательных реакций
Последствия разрушения полосатого тела
1. гипертонус скелетных мышц.
2. нарушение сложных двигательных реакций

Тип пораженных клеток
Полосатое тело на 96 процентов состоит из срединных шипиковых
Тип пораженных клеток
Полосатое тело на 96 процентов состоит из срединных шипиковых

HD
Normal
Уменьшение мозга на 30% при Хорее Гентингтона
HD
Normal
Уменьшение мозга на 30% при Хорее Гентингтона

Физические симптомы
Постепенное начало в возрасте 35-44 года;
Начальная стадия заболевания:
- резкие,
Физические симптомы
Постепенное начало в возрасте 35-44 года;
Начальная стадия заболевания:
- резкие,

Психические симптомы
Когнитивная сфера:
- поражаются способности управления деятельностью и поведением:
Психические симптомы
Когнитивная сфера:
- поражаются способности управления деятельностью и поведением:

Medication
Antipsychotics (hallucinations, delusions, violent outbursts): haloperidol, chlorpromazine, olanzapine (contraindicated if patient
Medication
Antipsychotics (hallucinations, delusions, violent outbursts): haloperidol, chlorpromazine, olanzapine (contraindicated if patient
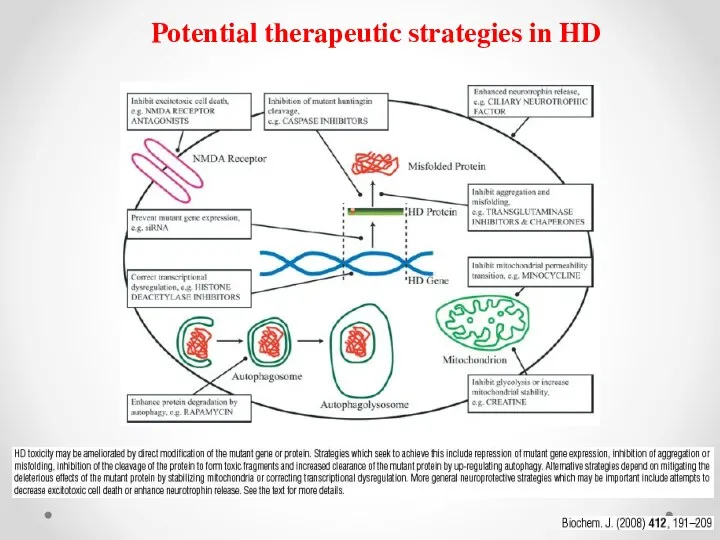
Potential therapeutic strategies in HD
Potential therapeutic strategies in HD

Болезнь Паркинсона
Болезнь Паркинсона

Болезнь Паркинсона
Вторая по распространенности среди нейродегенеративных болезней
α-синуклеин формирует амилоидные фибриллы
Накапливается в
Болезнь Паркинсона
Вторая по распространенности среди нейродегенеративных болезней
α-синуклеин формирует амилоидные фибриллы
Накапливается в

Болезнь Паркинсона - хроническое нейродегенеративное заболевание, связанное с нарушением деятельности базальных
Болезнь Паркинсона - хроническое нейродегенеративное заболевание, связанное с нарушением деятельности базальных

Распространенность
В России насчитывается до 350000 больных болезнью Паркинсона. В США болезнью Паркинсона
Распространенность
В России насчитывается до 350000 больных болезнью Паркинсона. В США болезнью Паркинсона

Эпидемиология болезни Паркинсона
В мире в целом насчитывается около 6 миллионов пациентов
Эпидемиология болезни Паркинсона
В мире в целом насчитывается около 6 миллионов пациентов

Причины болезни Паркинсона
Старение
Тот факт, что некоторые проявления болезни Паркинсона возникают и
Причины болезни Паркинсона
Старение
Тот факт, что некоторые проявления болезни Паркинсона возникают и
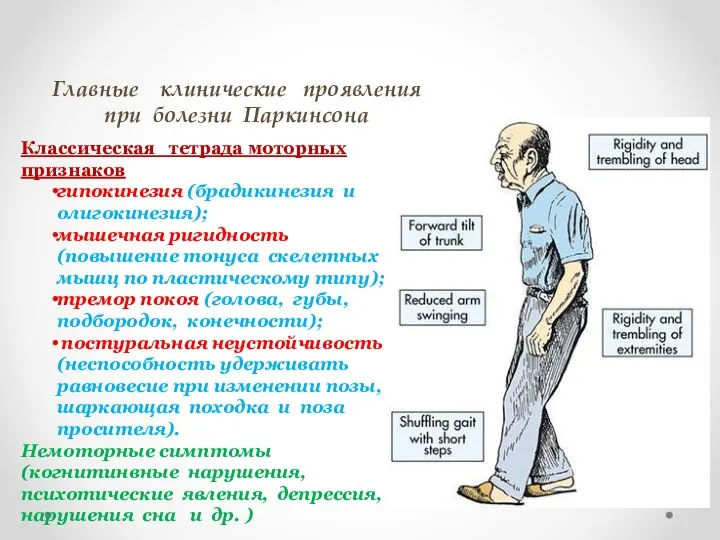
Главные клинические проявления
при болезни Паркинсона
Классическая тетрада моторных признаков
гипокинезия (брадикинезия
Главные клинические проявления
при болезни Паркинсона
Классическая тетрада моторных признаков
гипокинезия (брадикинезия
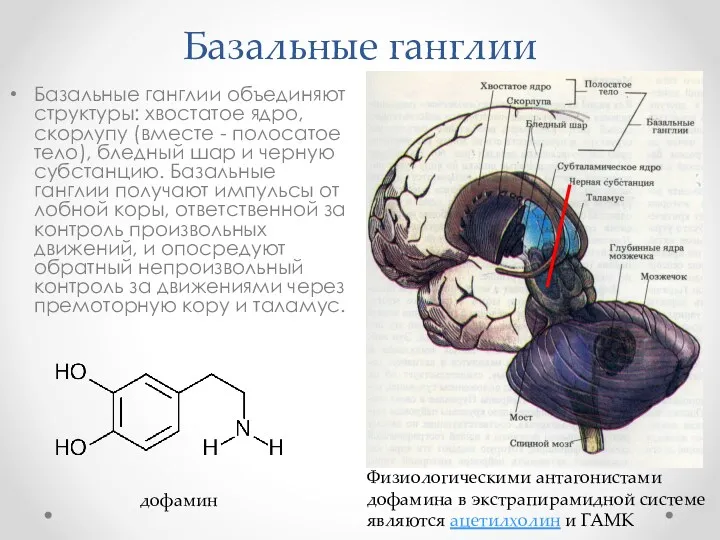
Базальные ганглии
Базальные ганглии объединяют структуры: хвостатое ядро, скорлупу (вместе - полосатое
Базальные ганглии
Базальные ганглии объединяют структуры: хвостатое ядро, скорлупу (вместе - полосатое
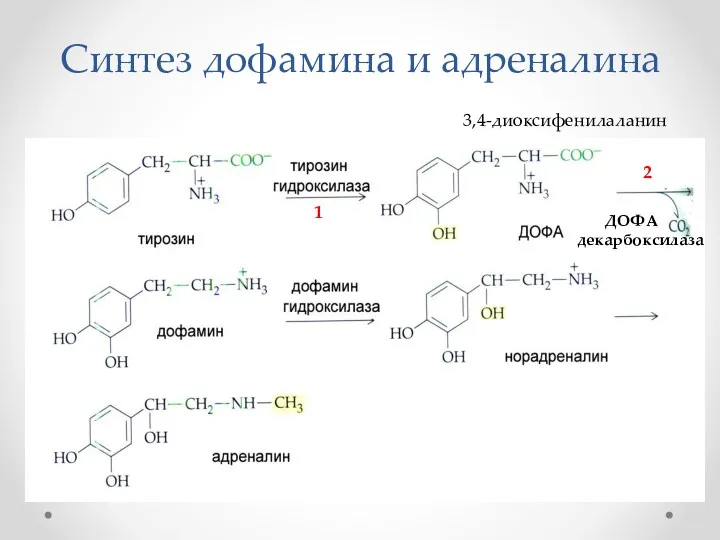
Синтез дофамина и адреналина
1
ДОФА декарбоксилаза
2
3,4-диоксифенилаланин
Синтез дофамина и адреналина
1
ДОФА декарбоксилаза
2
3,4-диоксифенилаланин

При болезни Паркинсона тельца Леви в первую очередь наблюдаются в области
При болезни Паркинсона тельца Леви в первую очередь наблюдаются в области
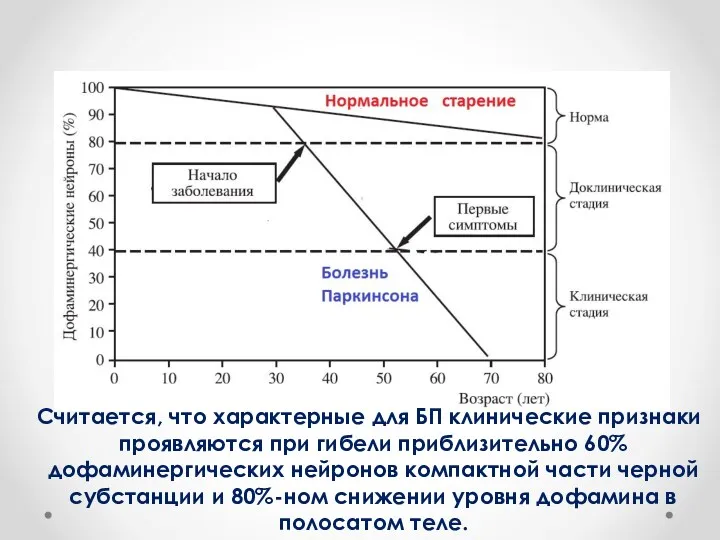
Cчитается, что характерные для БП клинические признаки проявляются при гибели
Cчитается, что характерные для БП клинические признаки проявляются при гибели
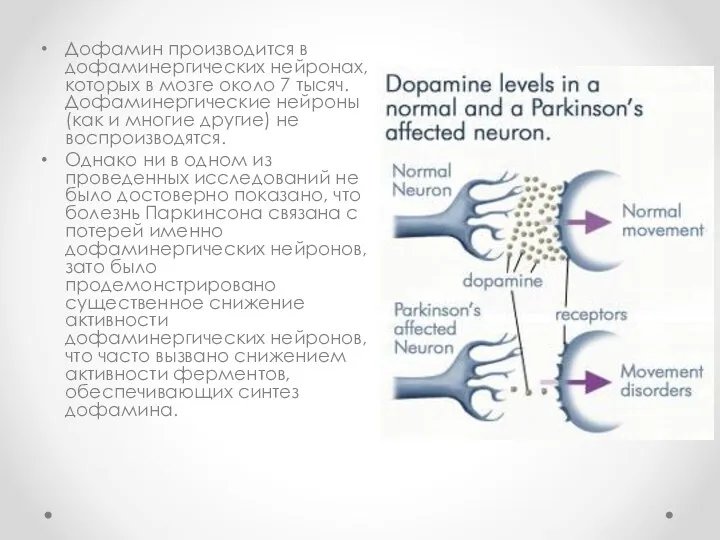
Дофамин производится в дофаминергических нейронах, которых в мозге около 7 тысяч.
Дофамин производится в дофаминергических нейронах, которых в мозге около 7 тысяч.
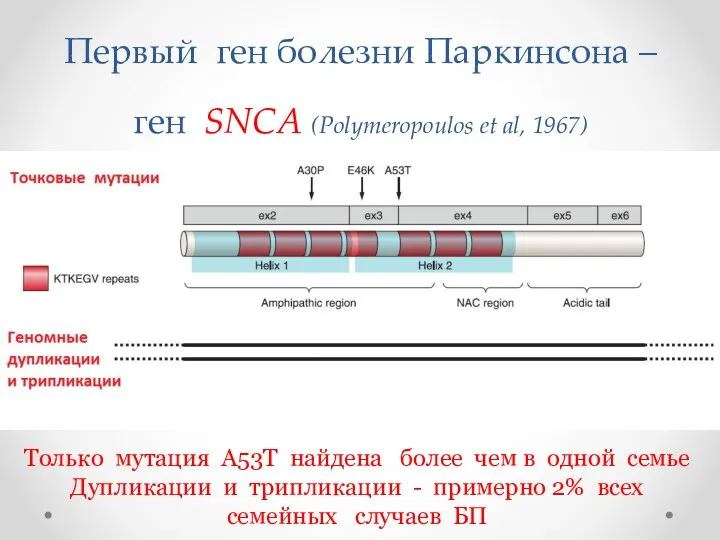
Первый ген болезни Паркинсона – ген SNCA (Polymeropoulos et al, 1967)
Только
Первый ген болезни Паркинсона – ген SNCA (Polymeropoulos et al, 1967)
Только
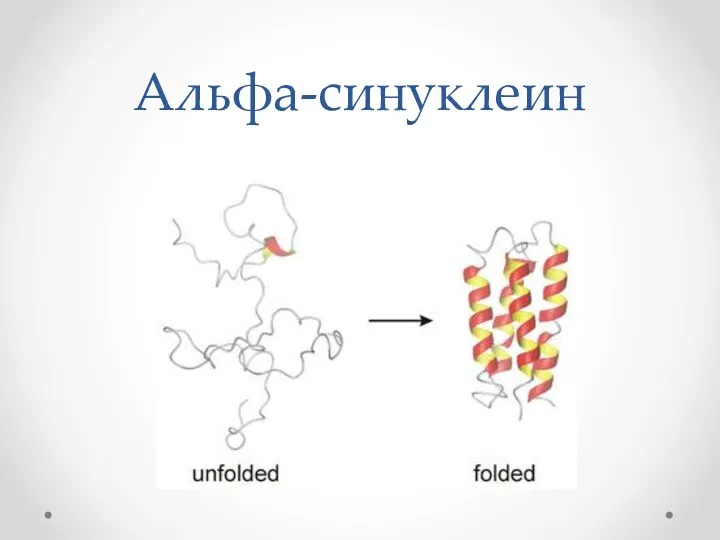
Альфа-синуклеин
Альфа-синуклеин
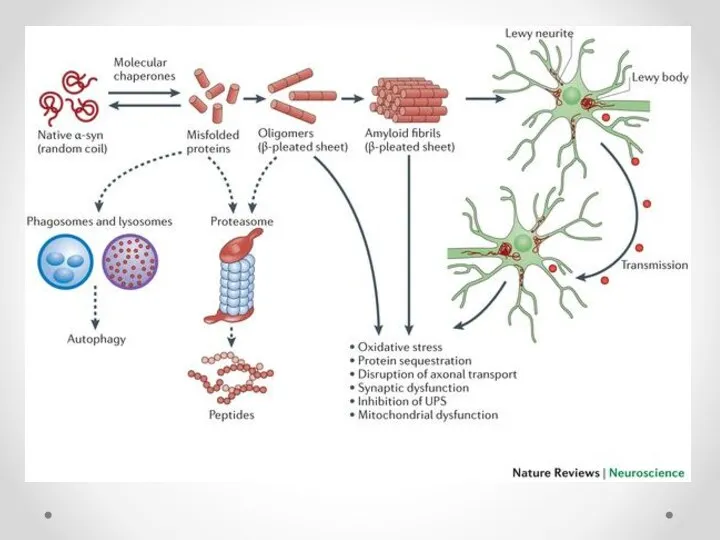
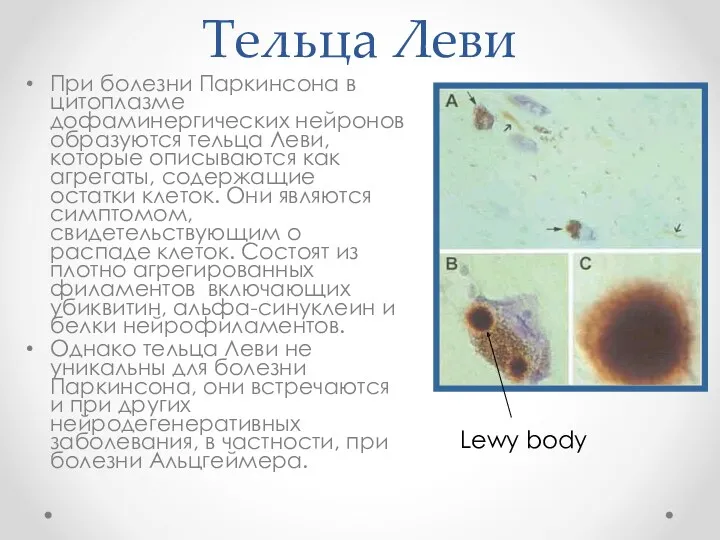
Тельца Леви
При болезни Паркинсона в цитоплазме дофаминергических нейронов образуются тельца Леви,
Тельца Леви
При болезни Паркинсона в цитоплазме дофаминергических нейронов образуются тельца Леви,
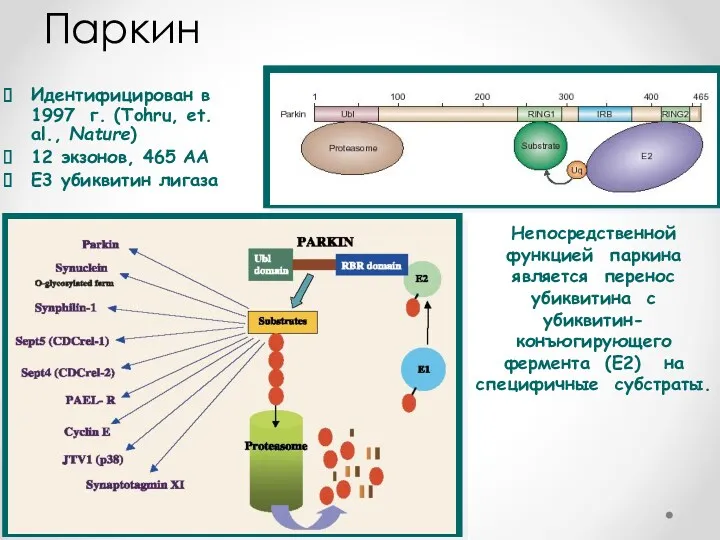
Идентифицирован в 1997 г. (Tohru, et. al., Nature)
12 экзонов, 465 AA
E3
Идентифицирован в 1997 г. (Tohru, et. al., Nature)
12 экзонов, 465 AA
E3

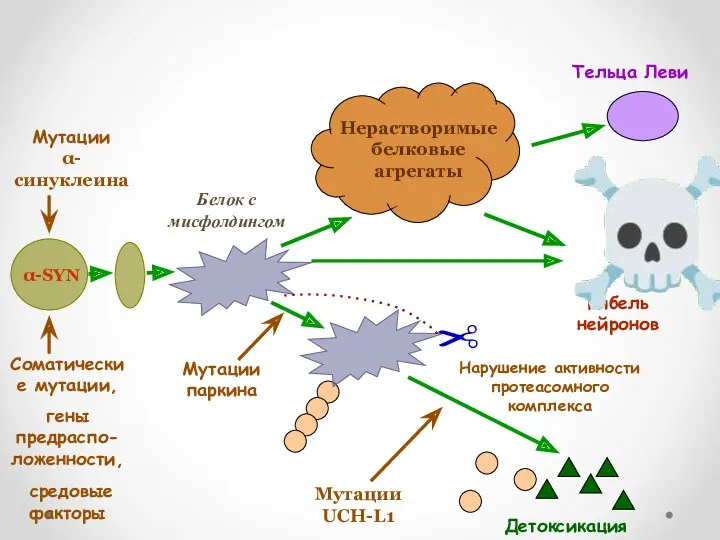
Мутации в белке-паркине
Показано, что белок паркин является важнейшим звеном системы клеточной
Мутации в белке-паркине
Показано, что белок паркин является важнейшим звеном системы клеточной
Белок с мисфолдингом
Белок с мисфолдингом

Гены, дефекты в которых приводят к БП
Гены транспорта и метаболизма дофамина
Гены, дефекты в которых приводят к БП
Гены транспорта и метаболизма дофамина

Связь между старением и болезнью Паркинсона
Исследуя мутации в митохондриальной ДНК (мтДНК) нейронов
Связь между старением и болезнью Паркинсона
Исследуя мутации в митохондриальной ДНК (мтДНК) нейронов

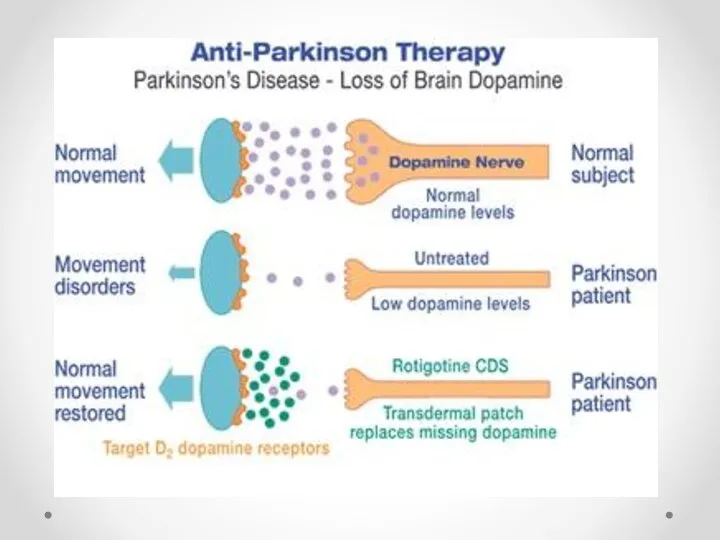
Лечение болезни Паркинсона
Средства с антиоксидантным эффектом (альфа-токоферол, тиоктовая кислота, десфероксамин, ингибиторы
Лечение болезни Паркинсона
Средства с антиоксидантным эффектом (альфа-токоферол, тиоктовая кислота, десфероксамин, ингибиторы
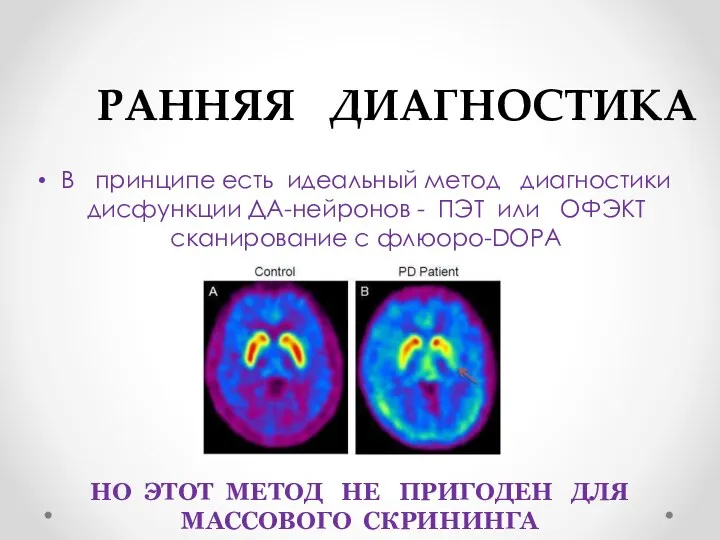
РАННЯЯ ДИАГНОСТИКА
В принципе есть идеальный метод диагностики дисфункции ДА-нейронов -
РАННЯЯ ДИАГНОСТИКА
В принципе есть идеальный метод диагностики дисфункции ДА-нейронов -

Массовый скрининг:
быстро - просто - относительно дешево
Нарушение обоняния
Массовый скрининг:
быстро - просто - относительно дешево
Нарушение обоняния

Роль диеты в профилактике нейродегенеративных заболеваний
Согласно результатам недавнего исследования, регулярное употребление
Роль диеты в профилактике нейродегенеративных заболеваний
Согласно результатам недавнего исследования, регулярное употребление
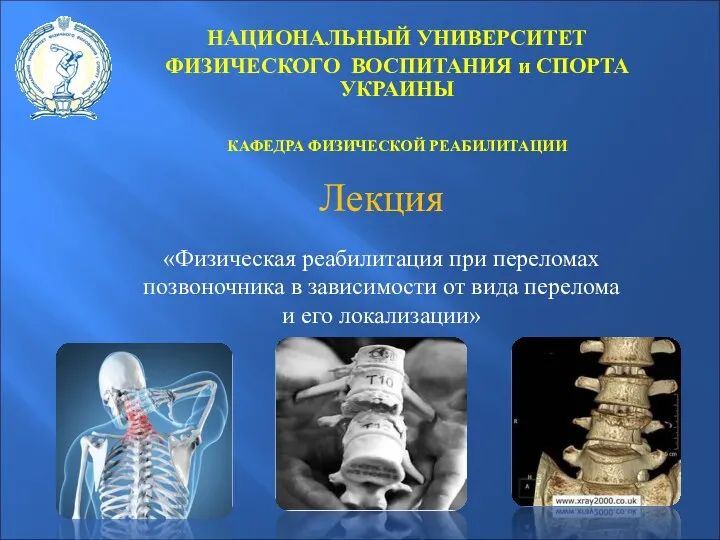 Физическая реабилитация при переломах позвоночника в зависимости от вида перелома и его локализации
Физическая реабилитация при переломах позвоночника в зависимости от вида перелома и его локализации Системная красная волчанка
Системная красная волчанка Новая классификация эпилептических приступов и эпилепсий. Разбор клинического случая
Новая классификация эпилептических приступов и эпилепсий. Разбор клинического случая Гигиенические требования к планировке и содержания детских и подростковых учреждениях
Гигиенические требования к планировке и содержания детских и подростковых учреждениях Пневмонии у детей (классификация, этиология, патогенез, клиника, осложнения, патогенетическая терапия )
Пневмонии у детей (классификация, этиология, патогенез, клиника, осложнения, патогенетическая терапия ) Гастроэнтеролог деонтологиясы
Гастроэнтеролог деонтологиясы Продукт добровольного медицинского страхования Клещевой энцефалит
Продукт добровольного медицинского страхования Клещевой энцефалит Воспалительные заболевания позвоночника. Хирургическое лечение
Воспалительные заболевания позвоночника. Хирургическое лечение Хирургия. Учебная литература
Хирургия. Учебная литература Вплив стресових факторів на організм людини
Вплив стресових факторів на організм людини Аномалии конституции (диатезы) у детей
Аномалии конституции (диатезы) у детей Іш сүзегі. Шигеллез
Іш сүзегі. Шигеллез Врожденная кишечная непроходимость
Врожденная кишечная непроходимость Кәсіби аурулар клиникасына кіріспе
Кәсіби аурулар клиникасына кіріспе Farmakologia_17_2023_LS_Zheludochnye_Lektsia
Farmakologia_17_2023_LS_Zheludochnye_Lektsia Лабораторная диагностика. Преаналитические требования. Интерпретация результатов ОАК
Лабораторная диагностика. Преаналитические требования. Интерпретация результатов ОАК Гемофилия. Гемофилия А этиологиясы және патогенезі
Гемофилия. Гемофилия А этиологиясы және патогенезі Жүрек қарыншалары мен жүрекшелері гипертрофиясының визуальді диагностика әдістері
Жүрек қарыншалары мен жүрекшелері гипертрофиясының визуальді диагностика әдістері Брюшной тиф
Брюшной тиф Уход за ребенком первого года жизни
Уход за ребенком первого года жизни Студенти-медики як конкурентне середовище
Студенти-медики як конкурентне середовище Аллергиялық жағдайлар кезінде дамитын шұғыл жағдайлардың диагностикалық тізбегі және жедел жәрдемі
Аллергиялық жағдайлар кезінде дамитын шұғыл жағдайлардың диагностикалық тізбегі және жедел жәрдемі Алиментарные заболевания
Алиментарные заболевания Патофизилогия системы дыхания
Патофизилогия системы дыхания Бесплодный брак
Бесплодный брак Сбор анамнеза стоматологического больного и описание локального статуса курируемого больного
Сбор анамнеза стоматологического больного и описание локального статуса курируемого больного Северо-восточный федеральный университет им. М.К. Аммосова. Медицинский институт
Северо-восточный федеральный университет им. М.К. Аммосова. Медицинский институт Развитие репродуктивной системы женщины
Развитие репродуктивной системы женщины