- Первичные иммунодефициты

Содержание
- 2. Определение понятия Это врожденные, генетически обусловленные иммунодефициты.
- 4. Классификация ПИДС Дефицит гуморального иммунитета: Сцепленная с Х-хромосомой агаммаглобулинемия (синдром Брутона) Общий вариабельный иммунодефицит Транзиторная гипогаммагобулинемия
- 7. Дефицит В-клеточного иммунитета
- 8. Болезнь Брутона болезнь, обусловленная дефектом гена хромосомы X, кодирующего синтез B-клеточной специфической тирозинкиназы Брутона (Btk)
- 9. Американский педиатр Огден Брутон впервые сделал описание этого заболевания в 1952 году. Это был мальчик, страдающий
- 10. Клиника До 9-месячного возраста младенцы защищены иммуноглобулинами матери и не болеют. Болеют только мальчики с тяжелыми
- 11. Лабораторная диагностика Двукратное снижение концентрации в крови всех типов иммуноглобулинов (IgG — менее 2 г/л, на
- 12. Лечение Внутривенные иммуноглобулины (ВИГ) вводят в суточной дозе 400 мг/кг в/в капельно по 1 мл/кг/час недоношенным
- 13. Селективный дефицит IgA — одна из обычных форм иммунодефицита, которая проявляется недостаточностью IgA в секретах. -
- 14. Клиника Рецидивирующие респираторные и желудочно-кишечные инфекции, атопические, аутоиммунные и онкологические заболевания. В некоторых случаях возможно отсутствие
- 15. Лабораторные исследования Снижение урованя IgA Анафилактические реакции на трасфузию Наличие в семейном анамнезе вариабельного неклассифицируемого иммунодефицита
- 16. Лечение Агрессивная антимикробная терапия и профилактика Агрессивная терапия атопических заболеваний Эффект от заместительной терапии в/в иммуноглобулином
- 17. Транзиторная гипогаммаглобулинемия у детей - Исчезновение IgG, переданного плоду через плаценту матери, вскоре после рождения, обычно
- 18. Клиника Рецидивирующие бактериальные синопульмонарные инфекции Рецидивирующие болезни ЖКТ Кандидоз Частые ОРВИ в раннем детском возрасте
- 19. Лабораторные исследования Уровень иммуноглобулинов ниже нормальных значений при сохранении продукции специфических АТ и клеточного иммунного ответа.
- 20. Лечение: Превентивная антибиотикотерапия Иногда – заместительная терапия гамма-глобулином
- 21. Общий вариабельный ИД - Один из наиболее распространенных ПИД, проявляющийся снижением уровня одних иммуноглобулинов при нормальном
- 22. Клиника Развиваются рецидивирующие инфекции респираторного и желудочно-кишечного трактов у мальчиков и девочек старше 2 лет. Характерна
- 23. Лабораторная диагностика Гипогаммаглобулинемия. ↓IgG, а также в большинстве случаев IgA и IgM ниже нормы при исключении
- 24. Лечение Заместительная терапия гамма-глобулином и антимикробная профилактика
- 25. Дефицит Т-клеточного звена иммунитета Составляет 5—10% общего количества первичных иммунодефицитов. Селективные дефекты клеточного звена иммунитета часто
- 26. Синдром Ди Джорджи В основе заболевания лежит гипоплазия или аплазия тимуса и паращитовидных желез Является результатом
- 27. Клиника У младенцев наблюдается низкое расположение ушных раковин, срединная расщелина лица, слабое недоразвитие нижней челюсти, гипертелоризм,
- 28. Неизмененная вилочковая железа у новорожденных в рентгеновском изображении на прямых рентгенограммах. Контуры железы показаны пунктиром
- 29. Диагностика снижение содержания в периферической крови Т-клеток (CD3+,CD4+,CD8+) Изменения в гуморальном звене иммунитета не всегда развиваются.
- 30. Лечение При полной аплазии тимуса трансплантация железы. При гипоплазии железы – назначают препараты тимуса. Лечение пороков
- 31. Хронический кожно-слизистый кандидоз Являются персистирующими или рецидивирующими инфекциями, обусловленными врожденными дефектами Т-клеток Наследование аутосомно-доминантное или рецессивное
- 32. Клиника Рецессивная форма: Аутоиммунные проявления Эндокринные проявления (гипотиреоз, недостаточность коры надпочечников, гипогонадизм, диабет) Мелкоочаговая алопеция Пернициозная
- 33. Диагностика нормальное содержании в периферической крови Т-лимфоцитов (CD3+), нормальная бласттрансформирующей способность на ФГА и пролиферативной активности
- 34. Лечение Назначается противомикозная терапия. Препараты, стимулирующие активность клеточного иммунитета.
- 35. Комбинированные иммунодефициты Общим для комбинированных иммунодефицитов является раннее их проявление, инфицирование множеством микроорганизмов, выраженная клиническая симптоматика,
- 36. Часто ПКИД сочетается с аномалиями скелета; для них характерна лимфоцитопения, снижение количества Т-лимфоцитов и их функциональной
- 37. Для ТКИД характерно: лимфоцитопения гипоплазия тимуса снижение количественного содержания Т- и В-лимфоцитов в крови и снижение
- 38. Лечение Единственным эффективным способом лечения таких болезней является трансплантация гемопоэтической ткани (костного мозга, клеток эмбриональной печени,
- 39. Синдром Луи-Барр Заболевание связано с дефектностью киназ, участвующих в регуляции клеточного цикла. Заболевание наследуется аутосомно-рецессивно. Дефект
- 40. Клиника Проявляется в возрасте от 5 мес до 3 лет Признаки мозжечковой атаксии, интенционный тремор, качание
- 41. Диагностика уменьшение содержания Т-лимфоцитов, сниженный уровень IgА, IgЕ, иногда IgG2, сниженный ответ в РБТ лимфоцитов на
- 42. Лечение Своевременная антибиотикотерапия при развитии инфекционных заболеваний. Ограждение ребенка от контактов с людьми, болеющими инфекционными заболеваниями
- 43. Синдром Вискотта-Олдрича комбинированная недостаточность В- и Т-лимфоцитов, которая характеризуется рецидивирующими инфекциями, экземой и тромбоцитопенией. Является сцепленным
- 44. Клиника В связи с нарушением функционирования Т- и В-лимфоцитов у пациентов развиваются инфекции, вызванные гноеродными бактериями
- 45. Диагностика Снижение количества и функции Т-клеток, повышении уровня IgE и IgA, низких уровнях IgM, и низких
- 46. Лечение Лечение заключается в пересадке костного мозга от HLA-идентичного донора. При значительном снижении гемоглобина проводят заместительную
- 47. Дефицит системы фагоцитов Синдром Чедиака-Хигаси редкое аутосомно-рецессивное заболевание, характеризующимся нарушением лизиса фагоцитированных бактерий, в результате чего
- 48. Он наследуется по аутосомно-рецессивному типу. Синдром вызван мутацией гена LYST (регулятор миграции лизосом; CHS1). В нейтрофилах
- 49. Клиника Клинические проявления включают альбинизм глаз и кожи, восприимчивость к респираторным и другим инфекциям. У 80%
- 50. Диагностика Обычно наблюдается нейтропения, снижение естественной цитотоксичности клеток-киллеров, и гипергаммаглобулинемия. Исследуются мазки периферической крови на наличие
- 51. Лечение Поддерживающая терапия с использованием антибиотиков, гамма-интерферона, в некоторых случаях кортикостероидов Пересадка костного мозга
- 52. Дефекты комплемента
- 53. Лечение и диагностика Общими принципами лечения этих заболеваний является введение «недостающих» ферментов, антибиотикотерапия, симптоматическое лечение. В
- 54. Интернет-источники http://immuninfo.ru/immunologiya/pervichnye-immunodeficity/deficit-sistemy-komplementa/ http://interferon.su/php/content.php?id=570&pr=print http://www.msdmanuals.com/ru/профессиональный/иммунология-аллергические-заболевания/иммунодефицитные-состояния
- 56. Скачать презентацию
Определение понятия
Это врожденные, генетически обусловленные иммунодефициты.
Определение понятия
Это врожденные, генетически обусловленные иммунодефициты.
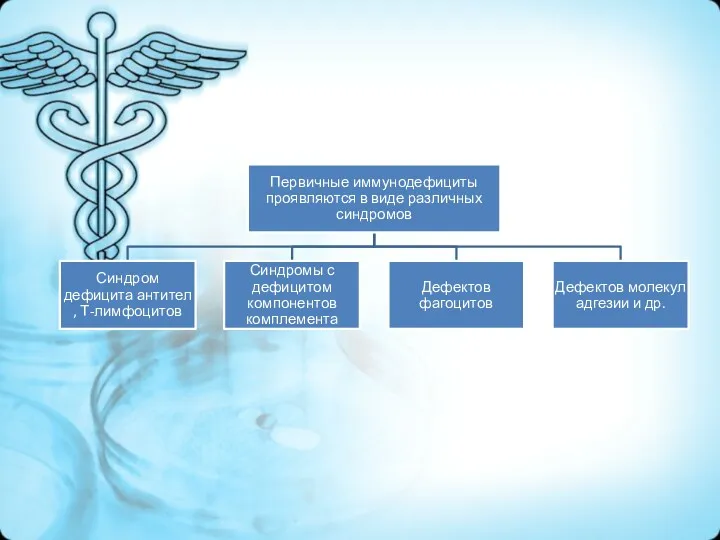

Классификация ПИДС
Дефицит гуморального иммунитета:
Сцепленная с Х-хромосомой агаммаглобулинемия (синдром Брутона)
Общий вариабельный иммунодефицит
Транзиторная
Классификация ПИДС
Дефицит гуморального иммунитета:
Сцепленная с Х-хромосомой агаммаглобулинемия (синдром Брутона)
Общий вариабельный иммунодефицит
Транзиторная
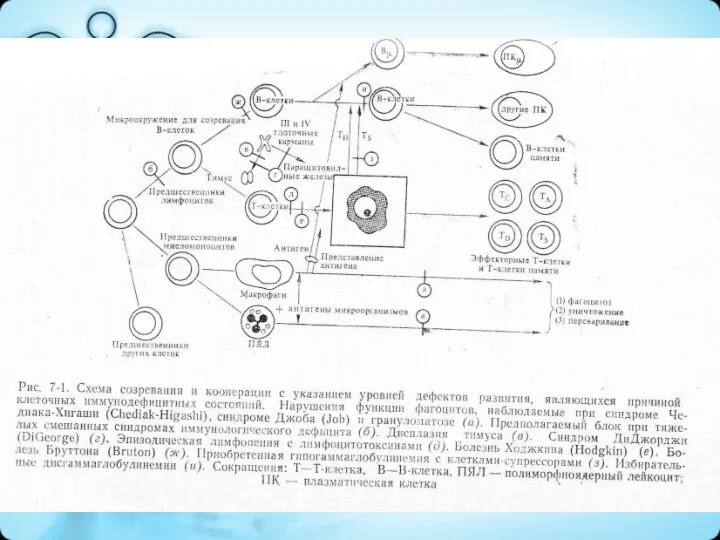

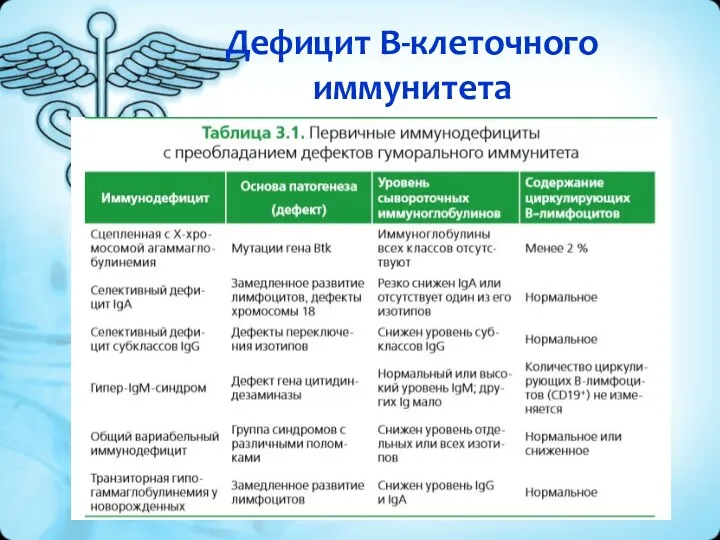
Дефицит В-клеточного иммунитета
Дефицит В-клеточного иммунитета
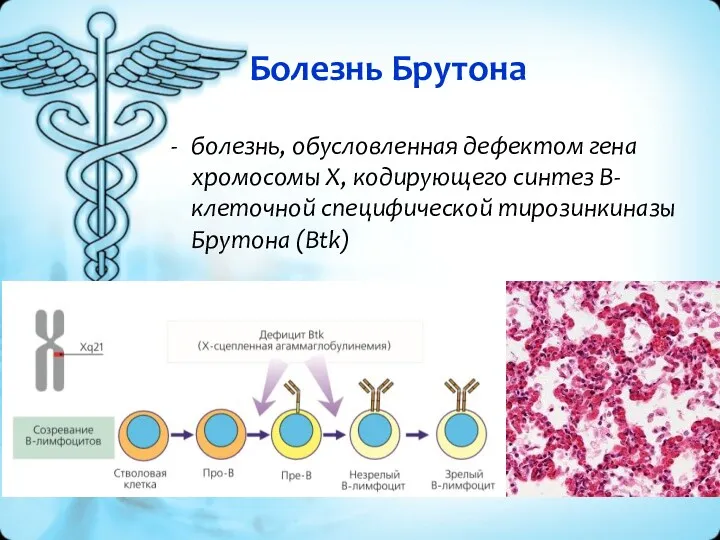
Болезнь Брутона
болезнь, обусловленная дефектом гена хромосомы X, кодирующего синтез B-клеточной специфической
Болезнь Брутона
болезнь, обусловленная дефектом гена хромосомы X, кодирующего синтез B-клеточной специфической
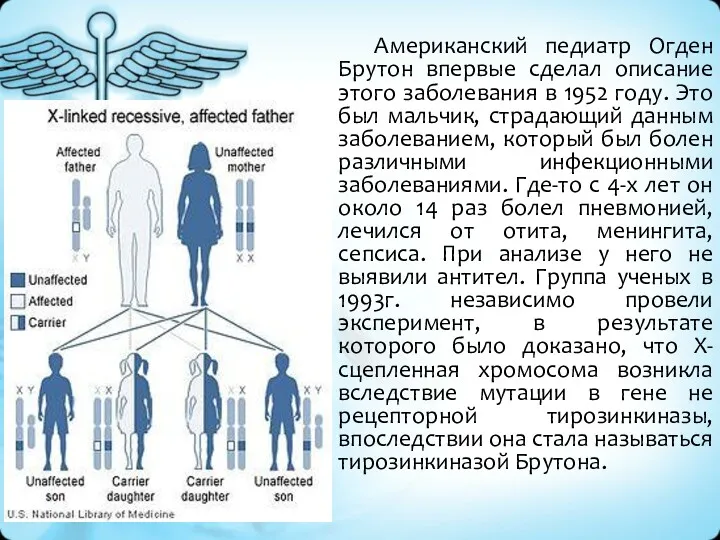
Американский педиатр Огден Брутон впервые сделал описание этого заболевания в 1952
Американский педиатр Огден Брутон впервые сделал описание этого заболевания в 1952
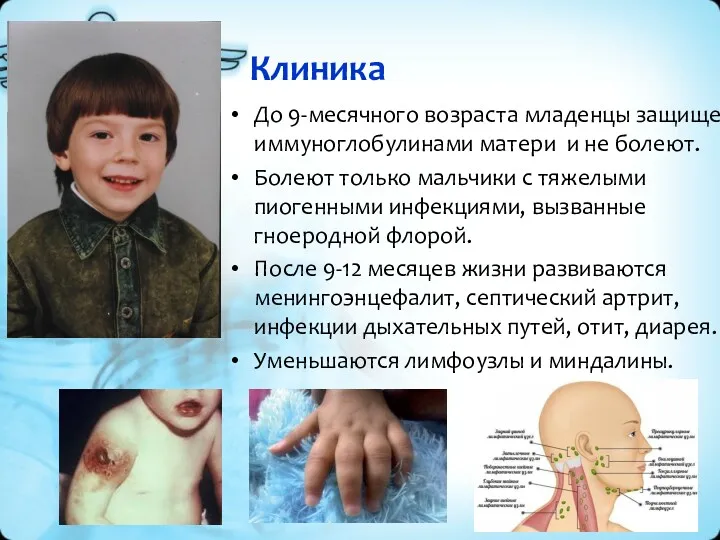
Клиника
До 9-месячного возраста младенцы защищены иммуноглобулинами матери и не болеют.
Болеют
Клиника
До 9-месячного возраста младенцы защищены иммуноглобулинами матери и не болеют.
Болеют

Лабораторная диагностика
Двукратное снижение концентрации в крови всех типов иммуноглобулинов (IgG —
Лабораторная диагностика
Двукратное снижение концентрации в крови всех типов иммуноглобулинов (IgG —
Лечение
Внутривенные иммуноглобулины (ВИГ) вводят в суточной дозе 400 мг/кг в/в капельно
Лечение
Внутривенные иммуноглобулины (ВИГ) вводят в суточной дозе 400 мг/кг в/в капельно

Селективный дефицит IgA
— одна из обычных форм иммунодефицита, которая проявляется недостаточностью
Селективный дефицит IgA
— одна из обычных форм иммунодефицита, которая проявляется недостаточностью
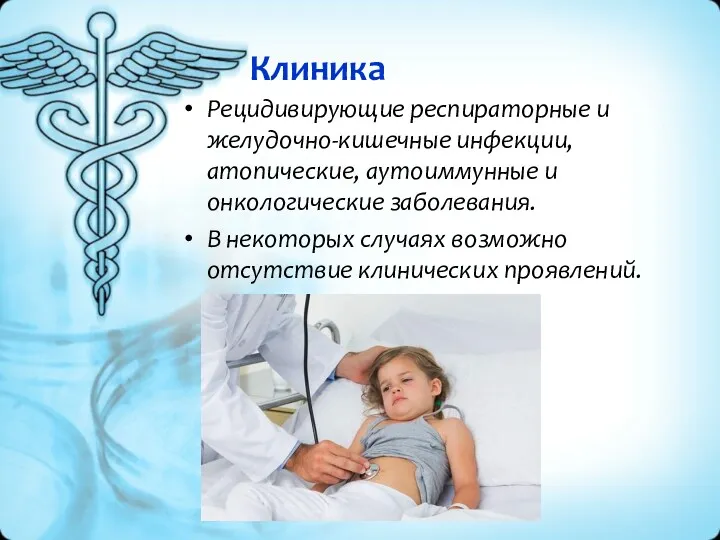
Клиника
Рецидивирующие респираторные и желудочно-кишечные инфекции, атопические, аутоиммунные и онкологические заболевания.
В некоторых
Клиника
Рецидивирующие респираторные и желудочно-кишечные инфекции, атопические, аутоиммунные и онкологические заболевания.
В некоторых

Лабораторные исследования
Снижение урованя IgA < 0,7 мг/л при нормальных показателях IgG
Лабораторные исследования
Снижение урованя IgA < 0,7 мг/л при нормальных показателях IgG

Лечение
Агрессивная антимикробная терапия и профилактика
Агрессивная терапия атопических заболеваний
Эффект от заместительной
Лечение
Агрессивная антимикробная терапия и профилактика
Агрессивная терапия атопических заболеваний
Эффект от заместительной
Транзиторная гипогаммаглобулинемия у детей
- Исчезновение IgG, переданного плоду через
Транзиторная гипогаммаглобулинемия у детей
- Исчезновение IgG, переданного плоду через
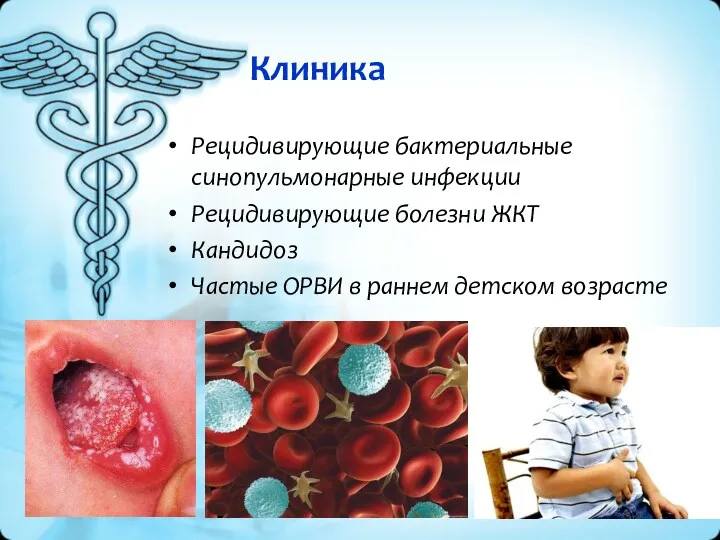
Клиника
Рецидивирующие бактериальные синопульмонарные инфекции
Рецидивирующие болезни ЖКТ
Кандидоз
Частые ОРВИ в раннем детском возрасте
Клиника
Рецидивирующие бактериальные синопульмонарные инфекции
Рецидивирующие болезни ЖКТ
Кандидоз
Частые ОРВИ в раннем детском возрасте

Лабораторные исследования
Уровень иммуноглобулинов ниже нормальных значений при сохранении продукции специфических АТ
Лабораторные исследования
Уровень иммуноглобулинов ниже нормальных значений при сохранении продукции специфических АТ
Лечение:
Превентивная антибиотикотерапия
Иногда – заместительная терапия гамма-глобулином
Лечение:
Превентивная антибиотикотерапия
Иногда – заместительная терапия гамма-глобулином
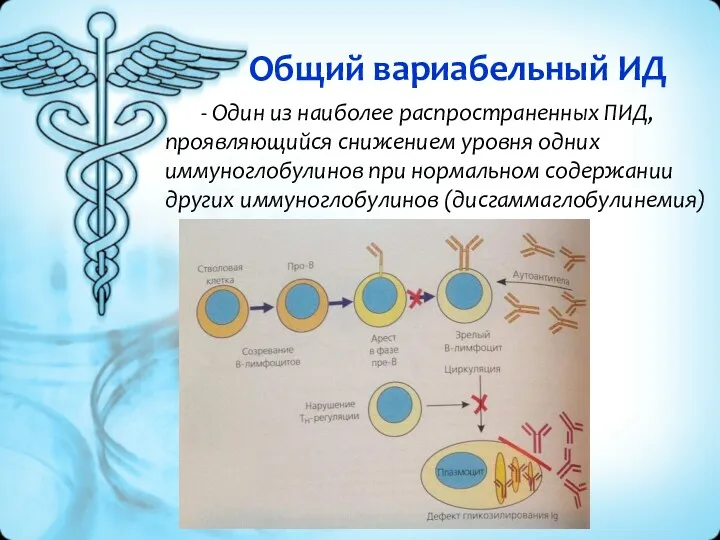
Общий вариабельный ИД
- Один из наиболее распространенных ПИД, проявляющийся снижением уровня
Общий вариабельный ИД
- Один из наиболее распространенных ПИД, проявляющийся снижением уровня
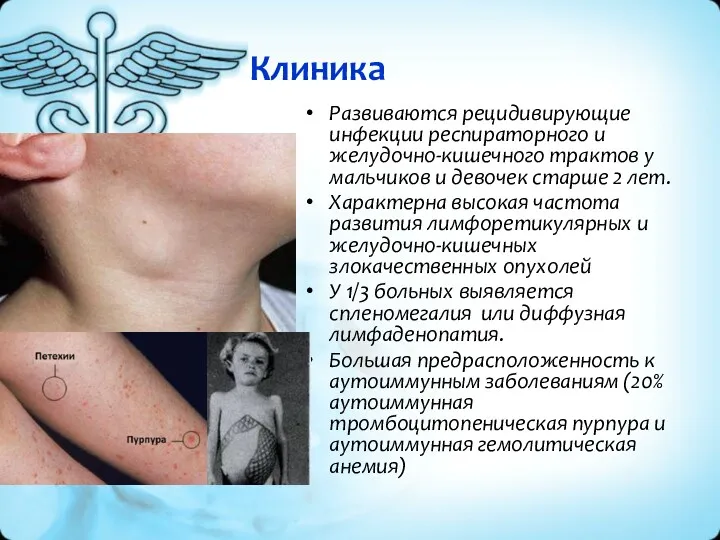
Клиника
Развиваются рецидивирующие инфекции респираторного и желудочно-кишечного трактов у мальчиков и
Клиника
Развиваются рецидивирующие инфекции респираторного и желудочно-кишечного трактов у мальчиков и

Лабораторная диагностика
Гипогаммаглобулинемия.
↓IgG, а также в большинстве случаев IgA и IgM ниже
Лабораторная диагностика
Гипогаммаглобулинемия.
↓IgG, а также в большинстве случаев IgA и IgM ниже
Лечение
Заместительная терапия гамма-глобулином и антимикробная профилактика
Лечение
Заместительная терапия гамма-глобулином и антимикробная профилактика

Дефицит Т-клеточного звена иммунитета
Составляет 5—10% общего количества первичных иммунодефицитов.
Селективные дефекты клеточного
Дефицит Т-клеточного звена иммунитета
Составляет 5—10% общего количества первичных иммунодефицитов.
Селективные дефекты клеточного
Синдром Ди Джорджи
В основе заболевания лежит гипоплазия или аплазия тимуса и
Синдром Ди Джорджи
В основе заболевания лежит гипоплазия или аплазия тимуса и

Клиника
У младенцев наблюдается низкое расположение ушных раковин, срединная расщелина лица,
Клиника
У младенцев наблюдается низкое расположение ушных раковин, срединная расщелина лица,
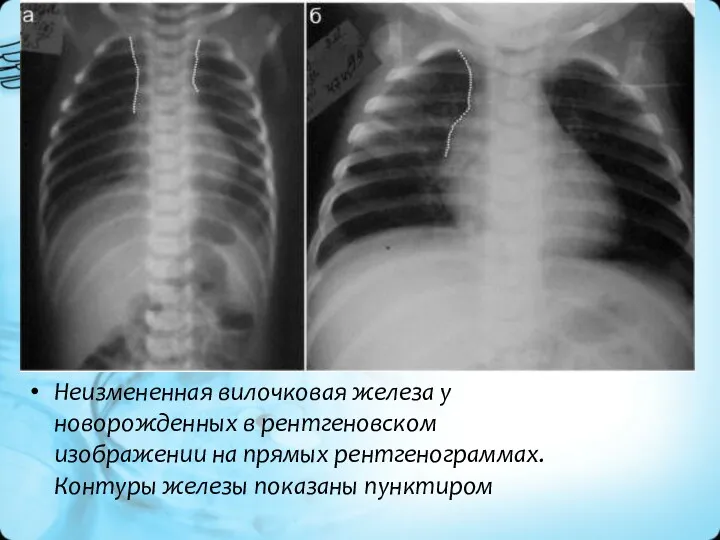
Неизмененная вилочковая железа у новорожденных в рентгеновском изображении на прямых рентгенограммах.
Неизмененная вилочковая железа у новорожденных в рентгеновском изображении на прямых рентгенограммах.

Диагностика
снижение содержания в периферической крови Т-клеток (CD3+,CD4+,CD8+)
Изменения в гуморальном звене
Диагностика
снижение содержания в периферической крови Т-клеток (CD3+,CD4+,CD8+)
Изменения в гуморальном звене

Лечение
При полной аплазии тимуса трансплантация железы.
При гипоплазии железы –
Лечение
При полной аплазии тимуса трансплантация железы.
При гипоплазии железы –

Хронический кожно-слизистый кандидоз
Являются персистирующими или рецидивирующими инфекциями, обусловленными врожденными дефектами Т-клеток
Наследование
Хронический кожно-слизистый кандидоз
Являются персистирующими или рецидивирующими инфекциями, обусловленными врожденными дефектами Т-клеток
Наследование
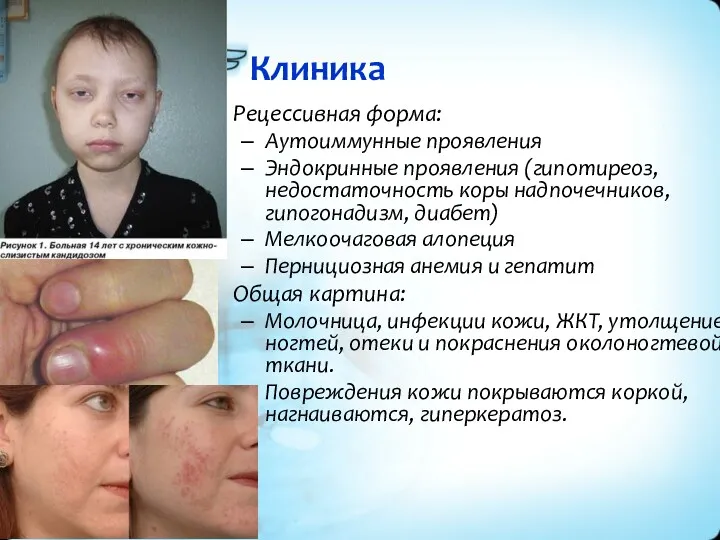
Клиника
Рецессивная форма:
Аутоиммунные проявления
Эндокринные проявления (гипотиреоз, недостаточность коры надпочечников, гипогонадизм, диабет)
Мелкоочаговая
Клиника
Рецессивная форма:
Аутоиммунные проявления
Эндокринные проявления (гипотиреоз, недостаточность коры надпочечников, гипогонадизм, диабет)
Мелкоочаговая

Диагностика
нормальное содержании в периферической крови Т-лимфоцитов (CD3+), нормальная бласттрансформирующей способность
Диагностика
нормальное содержании в периферической крови Т-лимфоцитов (CD3+), нормальная бласттрансформирующей способность
Лечение
Назначается противомикозная терапия.
Препараты, стимулирующие активность клеточного иммунитета.
Лечение
Назначается противомикозная терапия.
Препараты, стимулирующие активность клеточного иммунитета.

Комбинированные иммунодефициты
Общим для комбинированных иммунодефицитов является раннее их проявление, инфицирование
Комбинированные иммунодефициты
Общим для комбинированных иммунодефицитов является раннее их проявление, инфицирование

Часто ПКИД сочетается с аномалиями скелета; для них характерна лимфоцитопения, снижение
Часто ПКИД сочетается с аномалиями скелета; для них характерна лимфоцитопения, снижение

Для ТКИД характерно:
лимфоцитопения
гипоплазия тимуса
снижение количественного содержания Т- и В-лимфоцитов в
Для ТКИД характерно:
лимфоцитопения
гипоплазия тимуса
снижение количественного содержания Т- и В-лимфоцитов в

Лечение
Единственным эффективным способом лечения таких болезней является трансплантация гемопоэтической ткани
Лечение
Единственным эффективным способом лечения таких болезней является трансплантация гемопоэтической ткани
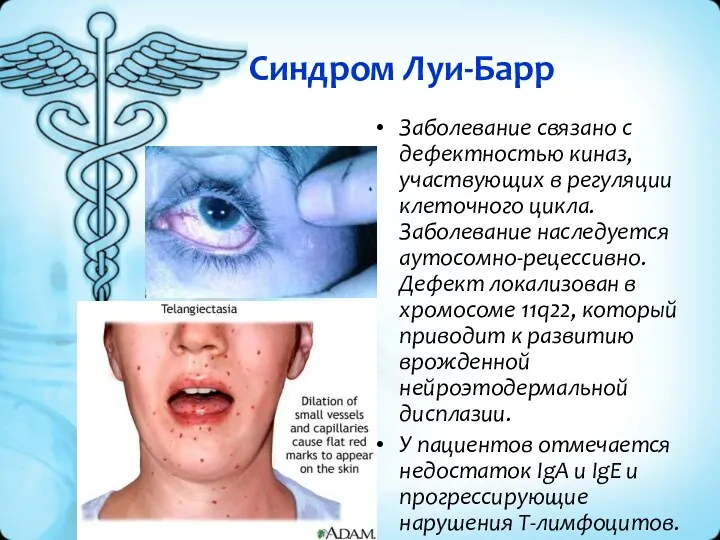
Синдром Луи-Барр
Заболевание связано с дефектностью киназ, участвующих в регуляции клеточного цикла.
Синдром Луи-Барр
Заболевание связано с дефектностью киназ, участвующих в регуляции клеточного цикла.
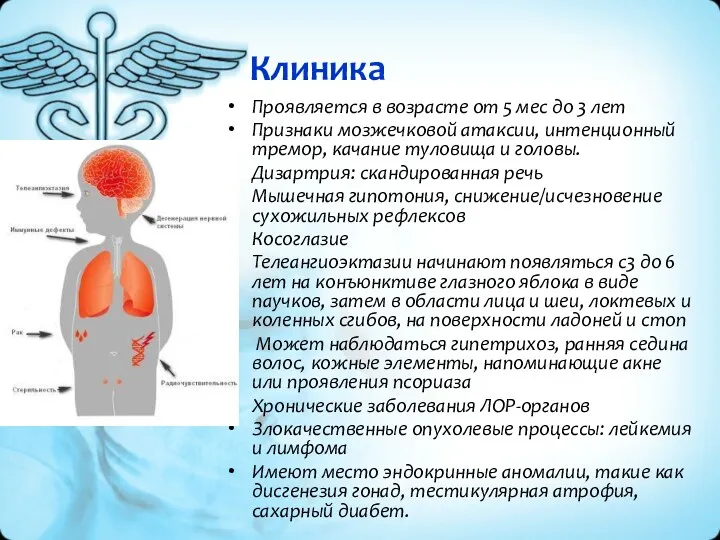
Клиника
Проявляется в возрасте от 5 мес до 3 лет
Признаки мозжечковой атаксии,
Клиника
Проявляется в возрасте от 5 мес до 3 лет
Признаки мозжечковой атаксии,

Диагностика
уменьшение содержания Т-лимфоцитов, сниженный уровень IgА, IgЕ, иногда IgG2, сниженный
Диагностика
уменьшение содержания Т-лимфоцитов, сниженный уровень IgА, IgЕ, иногда IgG2, сниженный

Лечение
Своевременная антибиотикотерапия при развитии инфекционных заболеваний.
Ограждение ребенка от контактов с людьми,
Лечение
Своевременная антибиотикотерапия при развитии инфекционных заболеваний.
Ограждение ребенка от контактов с людьми,

Синдром Вискотта-Олдрича
комбинированная недостаточность В- и Т-лимфоцитов, которая характеризуется рецидивирующими инфекциями, экземой
Синдром Вискотта-Олдрича
комбинированная недостаточность В- и Т-лимфоцитов, которая характеризуется рецидивирующими инфекциями, экземой
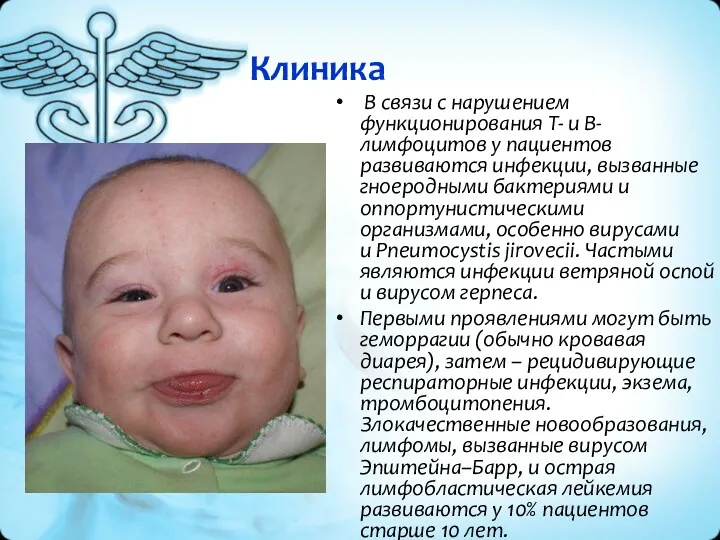
Клиника
В связи с нарушением функционирования Т- и В-лимфоцитов у пациентов
Клиника
В связи с нарушением функционирования Т- и В-лимфоцитов у пациентов

Диагностика
Снижение количества и функции Т-клеток, повышении уровня IgE и IgA, низких
Диагностика
Снижение количества и функции Т-клеток, повышении уровня IgE и IgA, низких

Лечение
Лечение заключается в пересадке костного мозга от HLA-идентичного донора.
При значительном
Лечение
Лечение заключается в пересадке костного мозга от HLA-идентичного донора.
При значительном
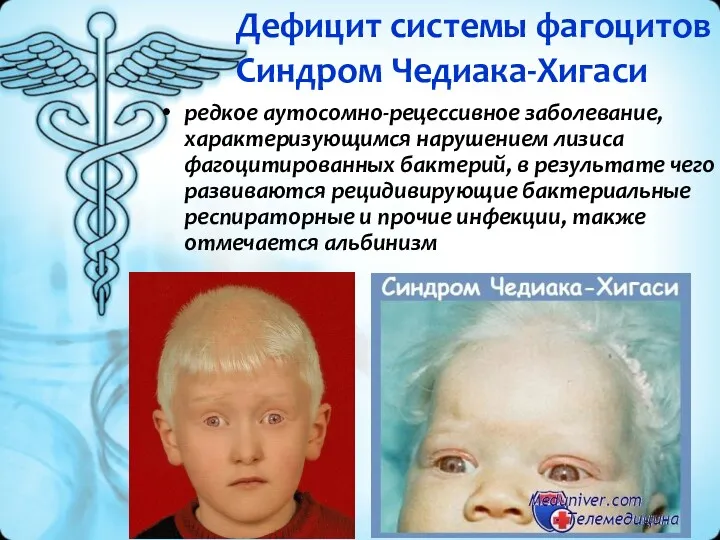
Дефицит системы фагоцитов
Синдром Чедиака-Хигаси
редкое аутосомно-рецессивное заболевание, характеризующимся нарушением лизиса
Дефицит системы фагоцитов
Синдром Чедиака-Хигаси
редкое аутосомно-рецессивное заболевание, характеризующимся нарушением лизиса
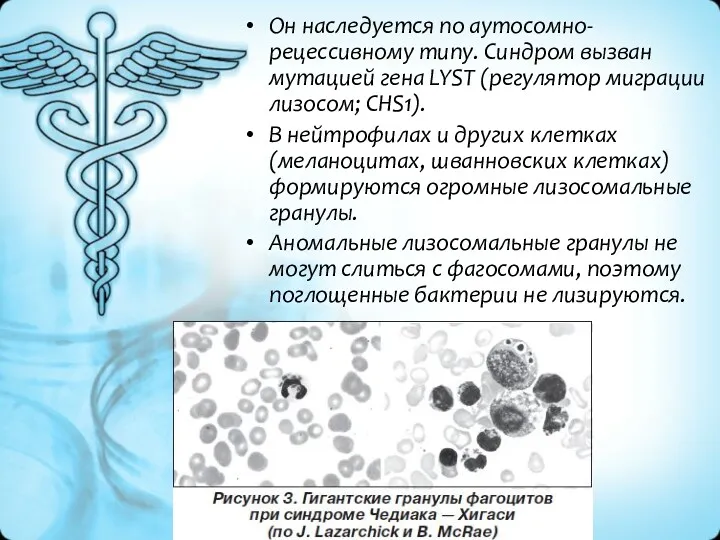
Он наследуется по аутосомно-рецессивному типу. Синдром вызван мутацией гена LYST (регулятор миграции лизосом; CHS1).
Он наследуется по аутосомно-рецессивному типу. Синдром вызван мутацией гена LYST (регулятор миграции лизосом; CHS1).

Клиника
Клинические проявления включают альбинизм глаз и кожи, восприимчивость к респираторным
Клиника
Клинические проявления включают альбинизм глаз и кожи, восприимчивость к респираторным

Диагностика
Обычно наблюдается нейтропения, снижение естественной цитотоксичности клеток-киллеров, и гипергаммаглобулинемия.
Исследуются
Диагностика
Обычно наблюдается нейтропения, снижение естественной цитотоксичности клеток-киллеров, и гипергаммаглобулинемия.
Исследуются

Лечение
Поддерживающая терапия с использованием антибиотиков, гамма-интерферона, в некоторых случаях кортикостероидов
Пересадка
Лечение
Поддерживающая терапия с использованием антибиотиков, гамма-интерферона, в некоторых случаях кортикостероидов
Пересадка
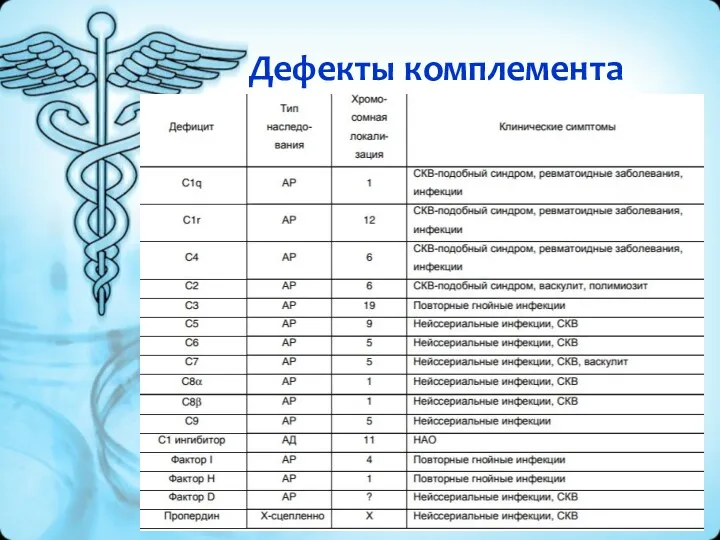
Дефекты комплемента
Дефекты комплемента

Лечение и диагностика
Общими принципами лечения этих заболеваний является введение «недостающих» ферментов,
Лечение и диагностика
Общими принципами лечения этих заболеваний является введение «недостающих» ферментов,

Интернет-источники
http://immuninfo.ru/immunologiya/pervichnye-immunodeficity/deficit-sistemy-komplementa/
http://interferon.su/php/content.php?id=570&pr=print
http://www.msdmanuals.com/ru/профессиональный/иммунология-аллергические-заболевания/иммунодефицитные-состояния
Интернет-источники
http://immuninfo.ru/immunologiya/pervichnye-immunodeficity/deficit-sistemy-komplementa/
http://interferon.su/php/content.php?id=570&pr=print
http://www.msdmanuals.com/ru/профессиональный/иммунология-аллергические-заболевания/иммунодефицитные-состояния
 Туберкулез қоздырғышы
Туберкулез қоздырғышы Теміртапшылықты анемия
Теміртапшылықты анемия Проблемы онкологии
Проблемы онкологии Балалардағы жедел синусит
Балалардағы жедел синусит Рассеянный склероз
Рассеянный склероз Эволюция хирургического лечения рака молочной железы
Эволюция хирургического лечения рака молочной железы Психогенные расстройства
Психогенные расстройства Щеплення. Профілактика щеплень
Щеплення. Профілактика щеплень Dentogene zysten nach WHO 2017
Dentogene zysten nach WHO 2017 Поранення магістральних судин кінцівок
Поранення магістральних судин кінцівок Іштің жабық жарақаты
Іштің жабық жарақаты Пороки развития органов мочеполовой системы
Пороки развития органов мочеполовой системы Вкладки. Классификация вкладок
Вкладки. Классификация вкладок Хронический холецистит. Классификация. Диагностика и лечение некалькулезного и калькулезного холецистита
Хронический холецистит. Классификация. Диагностика и лечение некалькулезного и калькулезного холецистита Адам ағзасының иммундық жүйесі
Адам ағзасының иммундық жүйесі Беременность и артериальная гипертензия
Беременность и артериальная гипертензия Спортивный массаж
Спортивный массаж Дәрілік өсімдіктер
Дәрілік өсімдіктер Сучасні стоматологічні цементи: цинк-фосфатні, полікарбоксилатні, скло-іономерні. Техніка пломбування скло-іономерними цементами
Сучасні стоматологічні цементи: цинк-фосфатні, полікарбоксилатні, скло-іономерні. Техніка пломбування скло-іономерними цементами Этические проблемы психиатрии. Нейроэтика. Лекция VIII
Этические проблемы психиатрии. Нейроэтика. Лекция VIII Посттрансфузионные реакции и осложения
Посттрансфузионные реакции и осложения Требования к условиям транспортирования и хранения иммунобиологических лекарственных препаратов
Требования к условиям транспортирования и хранения иммунобиологических лекарственных препаратов Кровоснабжение головного мозга. Виллизиев круг. Круг Захарченко
Кровоснабжение головного мозга. Виллизиев круг. Круг Захарченко Загальновійськовий набір пігулок. Попередження гіпотермії та виявлення ознак черепно-мозкової травми
Загальновійськовий набір пігулок. Попередження гіпотермії та виявлення ознак черепно-мозкової травми Алгоритм оказания первичной медико-санитарной помощи пациентам с ОРВИ и Covid-19
Алгоритм оказания первичной медико-санитарной помощи пациентам с ОРВИ и Covid-19 Экзогенная безлекарственная профилактика кариеса зубов в стоматологии
Экзогенная безлекарственная профилактика кариеса зубов в стоматологии Введение в анатомию
Введение в анатомию Жедел перитонит
Жедел перитонит