- Pheochromocytomas

Содержание
- 2. Pheochromocytomas rare, catecholamine-secreting, vascular, neuroendocrine tumors arising from chromaffin cells of the adrenal medulla ~80% extra-adrenal
- 3. Pheochromocytoma localization
- 4. Epidemiology rare cause of secondary hypertension less than 0.2% of patients with HTN incidence is approximately
- 5. Tumor characteristics ~ 95% of catecholamine-secreting tumors are in the abdomen 85-90% of which are intraadrenal
- 6. Clinical presentation The “classic triad”: episodic headache, sweating, and tachycardia – rarely seen Blood pressure: paroxysmal
- 7. PHEO may bee asymptomatic incidental imaging discovery (incidentaloma) genetic survey autopsy
- 8. Familial pheochromocytoma MEN 2 syndrome 95% autosomal dominant RET proto-oncogene mutation prevalence ~1/ 35,000 individuals ~
- 9. Familial pheochromocytoma MEN 2A (Sipple's syndrome) medullary thyroid cancer (MTC) in all patients, PHEO in 50%,
- 10. Familial pheochromocytoma MEN 2B (mucosal neuroma syndrome) MTC in all patients, PHEO in 50%, mucocutaneous neuromas,
- 11. Familial pheochromocytoma Neurofibromatosis type 1 prevalence ~ 1/ 3,000 individuals neurofibromas, multiple café-au-lait spots, axillary and
- 12. Familial pheochromocytoma von Hippel–Lindau disease (VHL) prevalence ~2–3/ 100,000 persons hemangioblastoma (cerebellum, spinal cord or brainstem),
- 13. Familial pheochromocytoma Familial paraganglioma syndromes Paraganglioma syndrome type 1-4 usually nonfunctional parasympathetic paragangliomas at skull base
- 14. Genetic vs. sporadic PHEO
- 15. When to suspect PHEO? Hyperadrenergic spells Resistant hypertension A familial syndrome that predisposes to PHEO (MEN2,
- 16. Catecholemine metabolism Metanephrine Normetanephrine Norepinephrine Vanillylmandelic acid COMT COMT MAO MAO
- 17. Pheochromocytoma diagnosis 24-hour urine collection for fractionated metanephrines and catecholamines Norepinephrine >170 mcg/24 h Epinephrine >35
- 18. Pheochromocytoma diagnosis Plasma fractionated metanephrines metanephrine normetanephrine high predictive value of a negative test high rate
- 20. Pheochromocytoma diagnosis 24-hour urinary vanillylmandelic acid (VMA) excretion poor diagnostic sensitivity and specificity Chromogranine A in
- 21. Pheochromocytoma imaging CT or MRI of the abdomen and pelvis Pheo Imaging characteristics • Usually large
- 22. Pheochromocytoma imaging a- FDG-PET b- abdominal CT c- DOTATATE-Scan
- 23. Pheochromocytoma treatment all patients should undergo a resection of the Pheo (laparascopic or open adrenalectomy) preoperative
- 25. Скачать презентацию

Pheochromocytomas
rare, catecholamine-secreting,
vascular, neuroendocrine tumors
arising from chromaffin cells of the
adrenal
Pheochromocytomas
rare, catecholamine-secreting,
vascular, neuroendocrine tumors
arising from chromaffin cells of the
adrenal
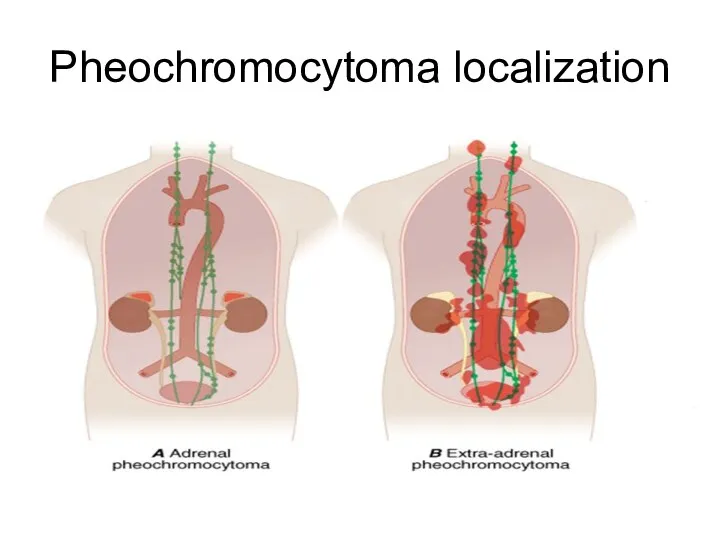
Pheochromocytoma localization
Pheochromocytoma localization

Epidemiology
rare cause of secondary hypertension
less than 0.2% of patients with
Epidemiology
rare cause of secondary hypertension
less than 0.2% of patients with

Tumor characteristics
~ 95% of catecholamine-secreting tumors are in the abdomen
85-90% of
Tumor characteristics
~ 95% of catecholamine-secreting tumors are in the abdomen
85-90% of

Clinical presentation
The “classic triad”: episodic headache, sweating, and tachycardia – rarely
Clinical presentation
The “classic triad”: episodic headache, sweating, and tachycardia – rarely

PHEO may bee asymptomatic
incidental imaging discovery (incidentaloma)
genetic survey
autopsy
PHEO may bee asymptomatic
incidental imaging discovery (incidentaloma)
genetic survey
autopsy

Familial pheochromocytoma
MEN 2 syndrome
95% autosomal dominant RET proto-oncogene mutation
prevalence ~1/
Familial pheochromocytoma
MEN 2 syndrome
95% autosomal dominant RET proto-oncogene mutation
prevalence ~1/
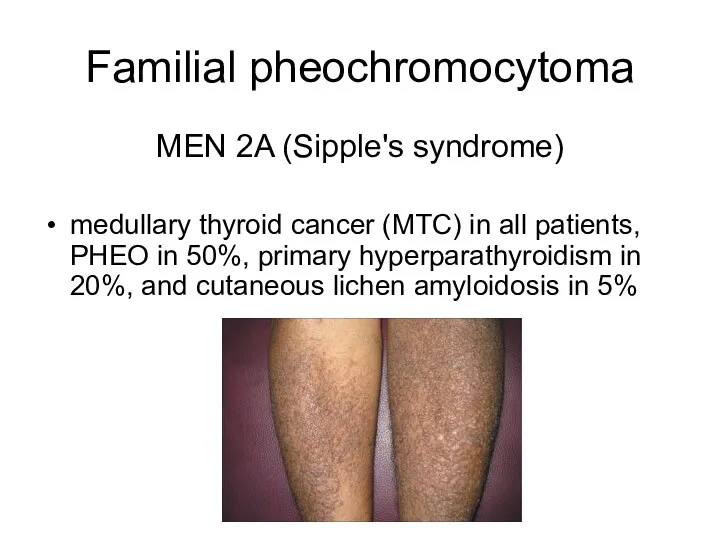
Familial pheochromocytoma
MEN 2A (Sipple's syndrome)
medullary thyroid cancer (MTC) in all
Familial pheochromocytoma
MEN 2A (Sipple's syndrome)
medullary thyroid cancer (MTC) in all

Familial pheochromocytoma
MEN 2B (mucosal neuroma syndrome)
MTC in all patients, PHEO in
Familial pheochromocytoma
MEN 2B (mucosal neuroma syndrome)
MTC in all patients, PHEO in
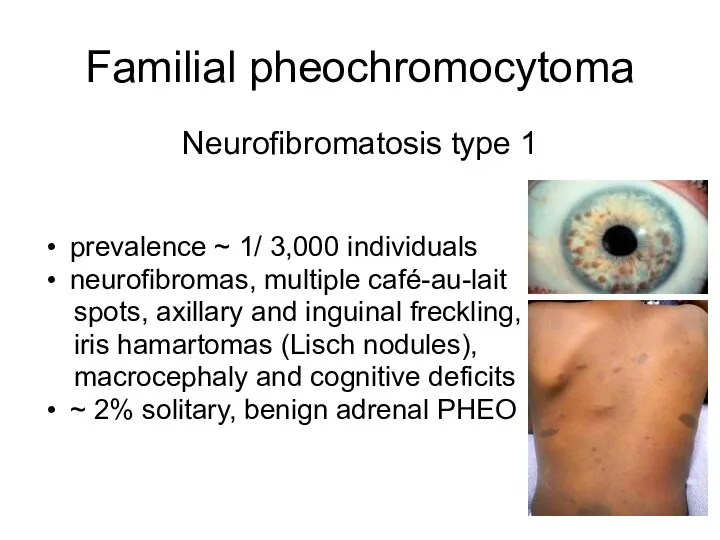
Familial pheochromocytoma
Neurofibromatosis type 1
prevalence ~ 1/ 3,000 individuals
neurofibromas, multiple café-au-lait
Familial pheochromocytoma
Neurofibromatosis type 1
prevalence ~ 1/ 3,000 individuals
neurofibromas, multiple café-au-lait

Familial pheochromocytoma
von Hippel–Lindau disease (VHL)
prevalence ~2–3/ 100,000 persons
hemangioblastoma (cerebellum, spinal
Familial pheochromocytoma
von Hippel–Lindau disease (VHL)
prevalence ~2–3/ 100,000 persons
hemangioblastoma (cerebellum, spinal

Familial pheochromocytoma
Familial paraganglioma syndromes
Paraganglioma syndrome type 1-4
usually nonfunctional parasympathetic paragangliomas
Familial pheochromocytoma
Familial paraganglioma syndromes
Paraganglioma syndrome type 1-4
usually nonfunctional parasympathetic paragangliomas
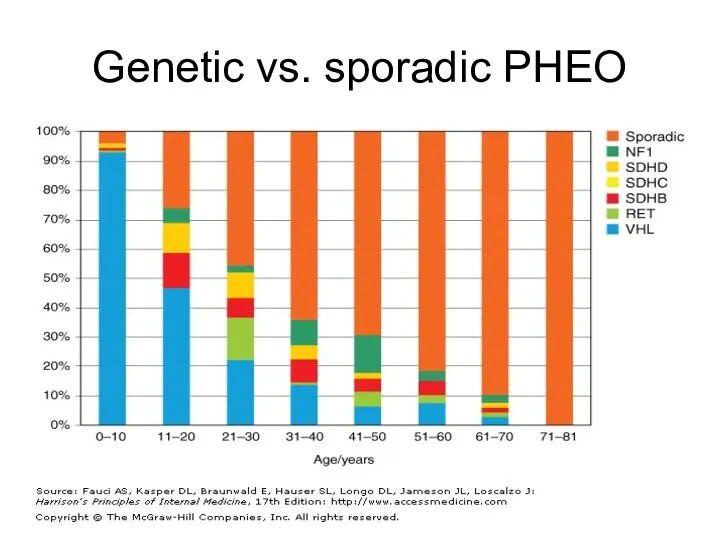
Genetic vs. sporadic PHEO
Genetic vs. sporadic PHEO

When to suspect PHEO?
Hyperadrenergic spells
Resistant hypertension
A familial syndrome that predisposes to
When to suspect PHEO?
Hyperadrenergic spells
Resistant hypertension
A familial syndrome that predisposes to
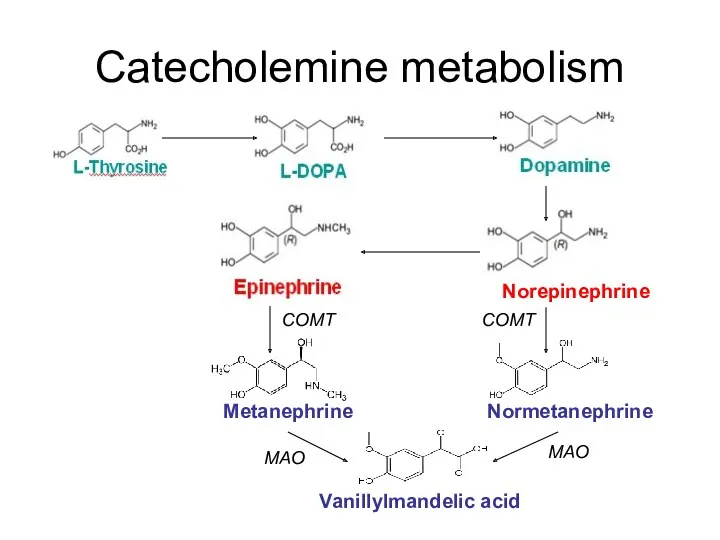
Catecholemine metabolism
Metanephrine
Normetanephrine
Norepinephrine
Vanillylmandelic acid
COMT
COMT
MAO
MAO
Catecholemine metabolism
Metanephrine
Normetanephrine
Norepinephrine
Vanillylmandelic acid
COMT
COMT
MAO
MAO

Pheochromocytoma diagnosis
24-hour urine collection for fractionated metanephrines and catecholamines
Norepinephrine >170 mcg/24
Pheochromocytoma diagnosis
24-hour urine collection for fractionated metanephrines and catecholamines
Norepinephrine >170 mcg/24

Pheochromocytoma diagnosis
Plasma fractionated metanephrines
metanephrine <0.3 nmol\l (fast), <0.5 nmo\l (non-fast)
normetanephrine <0.66
Pheochromocytoma diagnosis
Plasma fractionated metanephrines
metanephrine <0.3 nmol\l (fast), <0.5 nmo\l (non-fast)
normetanephrine <0.66


Pheochromocytoma diagnosis
24-hour urinary vanillylmandelic acid (VMA) excretion
poor diagnostic sensitivity
Pheochromocytoma diagnosis
24-hour urinary vanillylmandelic acid (VMA) excretion
poor diagnostic sensitivity

Pheochromocytoma imaging
CT or MRI of the abdomen and pelvis
Pheo Imaging characteristics
•
Pheochromocytoma imaging
CT or MRI of the abdomen and pelvis
Pheo Imaging characteristics
•
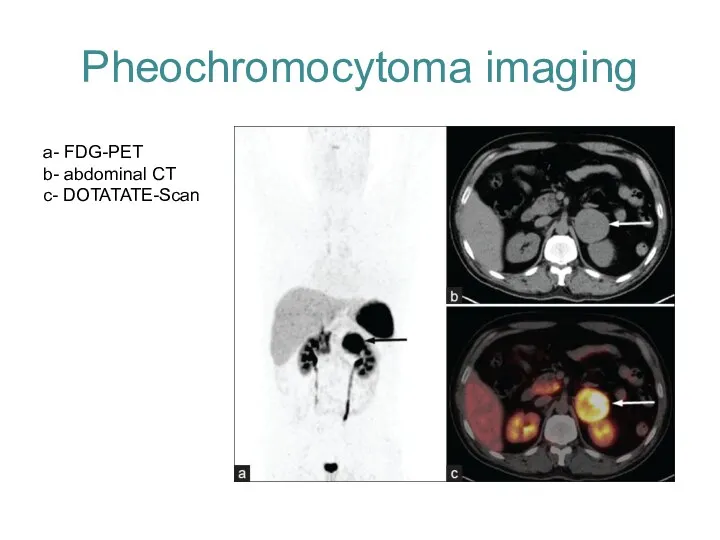
Pheochromocytoma imaging
a- FDG-PET
b- abdominal CT
c- DOTATATE-Scan
Pheochromocytoma imaging
a- FDG-PET
b- abdominal CT
c- DOTATATE-Scan

Pheochromocytoma treatment
all patients should undergo a resection of the Pheo (laparascopic
Pheochromocytoma treatment
all patients should undergo a resection of the Pheo (laparascopic
 Дорсопатия туралы түсінік
Дорсопатия туралы түсінік Ведение физиологической беременности. Принципы диспансеризации беременных
Ведение физиологической беременности. Принципы диспансеризации беременных Здоровые зубы
Здоровые зубы Особенности детей ОВЗ с умственной отсталостью. Обучение и воспитание
Особенности детей ОВЗ с умственной отсталостью. Обучение и воспитание Организация деятельности архива медицинской организации
Организация деятельности архива медицинской организации Гигиеническая оценка пищевых добавок, применяемых в РК
Гигиеническая оценка пищевых добавок, применяемых в РК Хвороби викликані вірусом
Хвороби викликані вірусом Терминальные состояние: стадии, клиника, диагностика, критерии оценки тяжести состояния больного
Терминальные состояние: стадии, клиника, диагностика, критерии оценки тяжести состояния больного Морфологія захворювань шлунково-кишкового тракту
Морфологія захворювань шлунково-кишкового тракту Нормативные требования к досье лекарственных средств
Нормативные требования к досье лекарственных средств Гестозы. Американская классификация гестозов
Гестозы. Американская классификация гестозов Идиопатический альвеолит. Синдром Хаммена-Рича
Идиопатический альвеолит. Синдром Хаммена-Рича Доброкачественные опухоли эпидермиса
Доброкачественные опухоли эпидермиса Лабораторное оборудование
Лабораторное оборудование Сравнительная характеристика методов лечения рака предстательной железы. Наиболее актуальные тактики ведения пациентов
Сравнительная характеристика методов лечения рака предстательной железы. Наиболее актуальные тактики ведения пациентов Пандемия испанского гриппа
Пандемия испанского гриппа Средства, влияющие на функции органов дыхания
Средства, влияющие на функции органов дыхания Исследование влияния физической и умственной нагрузки на утомляемость организма
Исследование влияния физической и умственной нагрузки на утомляемость организма Методы остановки кровотечения. Кровотечения и кровопотери
Методы остановки кровотечения. Кровотечения и кровопотери Шабуылдағы мотоатқыштар бригадасының медициналық қамтамасыз етілуін ұйымдастыру
Шабуылдағы мотоатқыштар бригадасының медициналық қамтамасыз етілуін ұйымдастыру Методы диагностики гиперчувствительности немедленного типа
Методы диагностики гиперчувствительности немедленного типа Основы ЭКГ
Основы ЭКГ Самай-төменгі жақ буынының анкилозы
Самай-төменгі жақ буынының анкилозы Строение тазобедренного сустава
Строение тазобедренного сустава Жиектер
Жиектер Наложение шин на руку
Наложение шин на руку Тифо-паратифозные заболевания. Брюшной тиф
Тифо-паратифозные заболевания. Брюшной тиф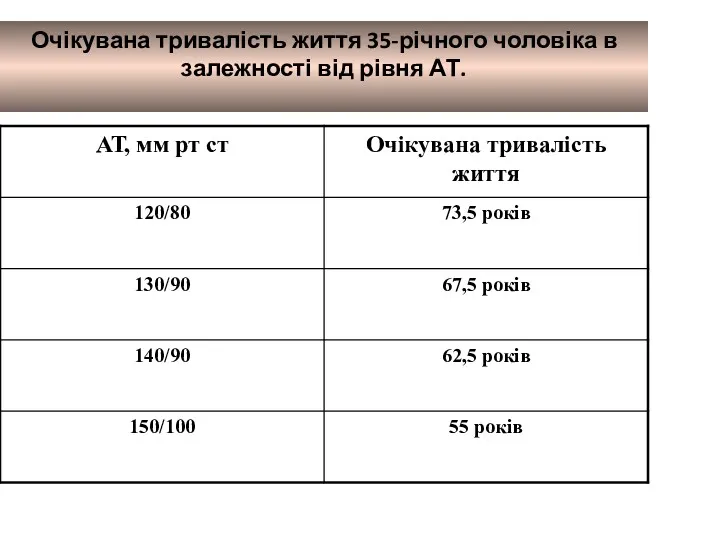 Очікувана тривалість життя 35-річного чоловіка в залежності від рівня АТ
Очікувана тривалість життя 35-річного чоловіка в залежності від рівня АТ