- Прионы и прионные заболевания

Содержание
- 2. ПЛАН Что такое прионы и прионные заболевания? Известные прионные заболевания Диагностика Профилактика Лечение
- 3. ЧТО ТАКОЕ ПРИОНЫ? Прио́ны (англ. prion от protein «белок» + infection «инфекция»— белки с аномальной третичной
- 4. Белок, вызывающий все известные прионные заболевания млекопитающих, называется PrP, сокращённое от PRion Protein. Его форма с
- 5. Основная особенность инфекционных прионов – это плотно упакованные β-листы, складчатые слои белковой структуры. Нормальные прионы встречаются
- 7. Прионы способны превращать конформацию гомологичных белков в подобную себе, тем самым увеличивая свою численность. По цепной
- 9. ПРИОННЫЕ БОЛЕЗНИ ЧЕЛОВЕКА болезнь Крейтцфельдта-Якоба синдром Герстмана-Штраусслера-Шейнкера болезнь куру фатальная семейная бессоница
- 10. БОЛЕЗНЬ КРЕЙТЦФЕЛЬДТА-ЯКОБА Прогрессирующее дистрофическое заболевание коры большого мозга, базальных ганглиев и спинного мозга. Считается основным проявлением
- 11. Накапливающиеся на поверхности клетки патологические белки блокируют процессы, происходящие на мембране, что приводит к гибели клетки.
- 12. быстро прогрессирующая — в течение 2 лет — («опустошающая») деменция с дезинтеграцией всех высших корковых функций;
- 14. Гистологический препарат — ткань лобной доли головного мозга больного болезнью Крейтцфельдта — Якоба
- 15. ФОРМЫ БОЛЕЗНИ Спонтанная Наследственная Ятрогенная Новый вариант – коровье бешенство
- 16. СИНДРОМ ГЕРСТМАНА — ШТРАУССЛЕРА — ШЕЙНКЕРА Синдром Герстмана — Штраусслера — Шейнкера (Gerstmann-Sträussler-Scheinker syndrome) — очень
- 17. Наблюдается атрофия мозжечка. Это очень типично для синдрома ГШШ Характеризуется мозжечковой атаксией, расстройствами глотания и фонации,
- 18. Фатальная семейная бессонница (англ. fatal familial insomnia, FFI) — редкое неизлечимое наследственное, нейро-дегенеративное (доминантнонаследуемое) прионное заболевание,
- 19. В кодоне 178 гена PRNP, находящегося в 20-й хромосоме, аспарагиновая кислота заменена на аспарагин. В результате
- 20. МЕТОДЫ ДИАГНОСТИКИ Исследование состава спинномозговой жидкости белковыми маркерами (ИФА, ИБ с моноклональными антителами) Выявление в крови
- 21. ЛЕЧЕНИЕ Только симптоматическое
- 23. Скачать презентацию

ПЛАН
Что такое прионы и прионные заболевания?
Известные прионные заболевания
Диагностика
Профилактика
Лечение
ПЛАН
Что такое прионы и прионные заболевания?
Известные прионные заболевания
Диагностика
Профилактика
Лечение

ЧТО ТАКОЕ ПРИОНЫ?
Прио́ны (англ. prion от protein «белок» + infection «инфекция»— белки с аномальной третичной структурой. Особый
ЧТО ТАКОЕ ПРИОНЫ?
Прио́ны (англ. prion от protein «белок» + infection «инфекция»— белки с аномальной третичной структурой. Особый

Белок, вызывающий все известные прионные заболевания млекопитающих, называется PrP, сокращённое от
Белок, вызывающий все известные прионные заболевания млекопитающих, называется PrP, сокращённое от

Основная особенность инфекционных прионов – это плотно упакованные β-листы, складчатые слои
Основная особенность инфекционных прионов – это плотно упакованные β-листы, складчатые слои
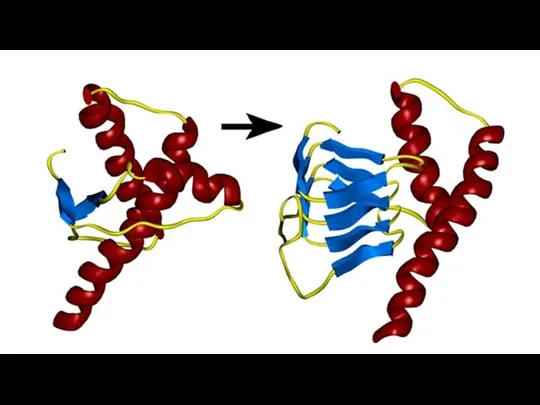

Прионы способны превращать конформацию гомологичных белков в подобную себе, тем самым
Прионы способны превращать конформацию гомологичных белков в подобную себе, тем самым

ПРИОННЫЕ БОЛЕЗНИ ЧЕЛОВЕКА
болезнь Крейтцфельдта-Якоба
синдром Герстмана-Штраусслера-Шейнкера
болезнь куру
фатальная семейная
ПРИОННЫЕ БОЛЕЗНИ ЧЕЛОВЕКА
болезнь Крейтцфельдта-Якоба
синдром Герстмана-Штраусслера-Шейнкера
болезнь куру
фатальная семейная

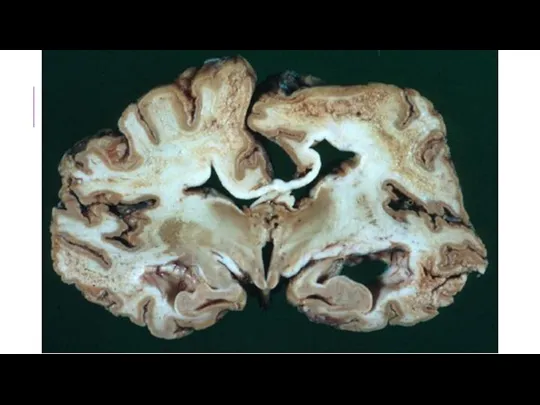
БОЛЕЗНЬ КРЕЙТЦФЕЛЬДТА-ЯКОБА
Прогрессирующее дистрофическое заболевание коры большого мозга, базальных ганглиев и спинного мозга. Считается основным проявлением губчатой
БОЛЕЗНЬ КРЕЙТЦФЕЛЬДТА-ЯКОБА
Прогрессирующее дистрофическое заболевание коры большого мозга, базальных ганглиев и спинного мозга. Считается основным проявлением губчатой

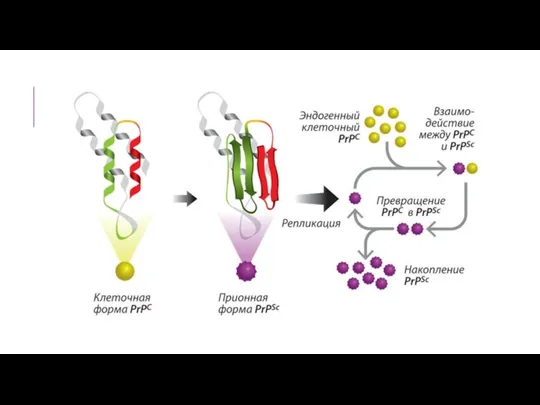
Накапливающиеся на поверхности клетки патологические белки блокируют процессы, происходящие на мембране,
Накапливающиеся на поверхности клетки патологические белки блокируют процессы, происходящие на мембране,

быстро прогрессирующая — в течение 2 лет — («опустошающая») деменция с дезинтеграцией всех высших корковых
быстро прогрессирующая — в течение 2 лет — («опустошающая») деменция с дезинтеграцией всех высших корковых
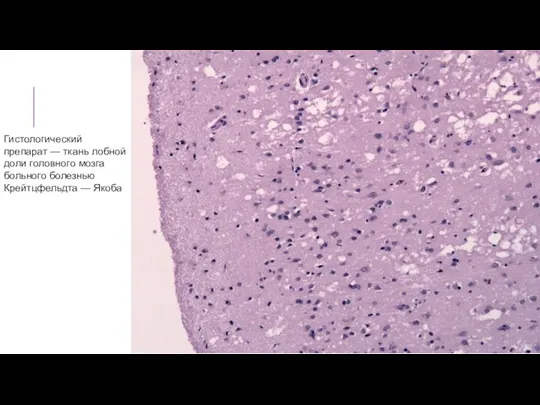
Гистологический препарат — ткань лобной доли головного мозга больного болезнью Крейтцфельдта —
Гистологический препарат — ткань лобной доли головного мозга больного болезнью Крейтцфельдта —

ФОРМЫ БОЛЕЗНИ
Спонтанная
Наследственная
Ятрогенная
Новый вариант – коровье бешенство
ФОРМЫ БОЛЕЗНИ
Спонтанная
Наследственная
Ятрогенная
Новый вариант – коровье бешенство

СИНДРОМ ГЕРСТМАНА — ШТРАУССЛЕРА — ШЕЙНКЕРА
Синдром Герстмана — Штраусслера — Шейнкера (Gerstmann-Sträussler-Scheinker syndrome) — очень редкое, обычно
СИНДРОМ ГЕРСТМАНА — ШТРАУССЛЕРА — ШЕЙНКЕРА
Синдром Герстмана — Штраусслера — Шейнкера (Gerstmann-Sträussler-Scheinker syndrome) — очень редкое, обычно
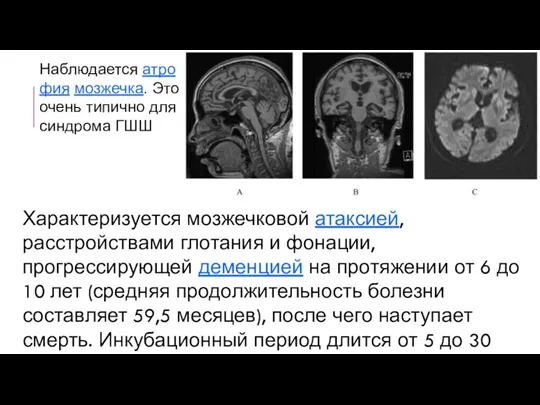
Наблюдается атрофия мозжечка. Это очень типично для синдрома ГШШ
Характеризуется мозжечковой атаксией, расстройствами глотания и
Наблюдается атрофия мозжечка. Это очень типично для синдрома ГШШ
Характеризуется мозжечковой атаксией, расстройствами глотания и

Фатальная семейная бессонница (англ. fatal familial insomnia, FFI) — редкое неизлечимое наследственное, нейро-дегенеративное (доминантнонаследуемое) прионное заболевание,
Фатальная семейная бессонница (англ. fatal familial insomnia, FFI) — редкое неизлечимое наследственное, нейро-дегенеративное (доминантнонаследуемое) прионное заболевание,

В кодоне 178 гена PRNP, находящегося в 20-й хромосоме, аспарагиновая кислота заменена на аспарагин. В результате форма белковой молекулы изменяется,
В кодоне 178 гена PRNP, находящегося в 20-й хромосоме, аспарагиновая кислота заменена на аспарагин. В результате форма белковой молекулы изменяется,

МЕТОДЫ ДИАГНОСТИКИ
Исследование состава спинномозговой жидкости белковыми маркерами (ИФА, ИБ с моноклональными
МЕТОДЫ ДИАГНОСТИКИ
Исследование состава спинномозговой жидкости белковыми маркерами (ИФА, ИБ с моноклональными

ЛЕЧЕНИЕ
Только симптоматическое
ЛЕЧЕНИЕ
Только симптоматическое
 Артрит и артроз. Причины, симптомы, осложнения, лечение
Артрит и артроз. Причины, симптомы, осложнения, лечение Моделировочные материлы в ортопедической стоматологии
Моделировочные материлы в ортопедической стоматологии Микроэлементы и их роль в организме человека
Микроэлементы и их роль в организме человека Обучение пациента правилам диабетического режима. Определение гликемии и применение инсулина методов введения в школе диабета
Обучение пациента правилам диабетического режима. Определение гликемии и применение инсулина методов введения в школе диабета Дивертикулярная болезнь ободочной кишки у пожилых
Дивертикулярная болезнь ободочной кишки у пожилых Антибактериальные химиотерапевтические средства
Антибактериальные химиотерапевтические средства Техника омоложения кожи
Техника омоложения кожи Бронхиалдық астма-инфекциялық аллергиялық ауру
Бронхиалдық астма-инфекциялық аллергиялық ауру Болезнь Иценко-Кушинга. Клинический случай
Болезнь Иценко-Кушинга. Клинический случай Дизартрия. Симптомы дизартрии
Дизартрия. Симптомы дизартрии Врожденные пороки сердца: педиатрические аспекты
Врожденные пороки сердца: педиатрические аспекты Туберкулез и его профилактика
Туберкулез и его профилактика Внутриутробные инфекции плода и новорожденного. Пути инфицирования. Этиология. Патогенез. Клиника. Диагностика
Внутриутробные инфекции плода и новорожденного. Пути инфицирования. Этиология. Патогенез. Клиника. Диагностика Железодефицитные анемии у новорожденных современные возможности профилактики и лечения
Железодефицитные анемии у новорожденных современные возможности профилактики и лечения Механизмы врожденного иммунитета
Механизмы врожденного иммунитета Острые лейкозы
Острые лейкозы Жақ - бет аймағының одонтогенді емес ісіктері
Жақ - бет аймағының одонтогенді емес ісіктері Хвороби надниркових залоз і гіпофіза. Єтіологія, Патогенез
Хвороби надниркових залоз і гіпофіза. Єтіологія, Патогенез Фармацевтің ақпараттық және кеңестік қызметі
Фармацевтің ақпараттық және кеңестік қызметі Санкт-Петербургская олимпиада по оказанию первой помощи
Санкт-Петербургская олимпиада по оказанию первой помощи Нейровизуализационные методы исследование нервной системы
Нейровизуализационные методы исследование нервной системы Синдром красного глаза
Синдром красного глаза Жатыр мойнының қатерлі ісігі жүктілік кезінде
Жатыр мойнының қатерлі ісігі жүктілік кезінде Добро пожаловать на День здорового образа жизни
Добро пожаловать на День здорового образа жизни Ожирение. Этиопатогенез ожирения
Ожирение. Этиопатогенез ожирения Денсаулық сақтау жүйесі және медициналық көмекті ұйымдастыру
Денсаулық сақтау жүйесі және медициналық көмекті ұйымдастыру Топографическая анатомия. Инструменты
Топографическая анатомия. Инструменты Телемедицина - использование компьютерных и телекоммуникационных технологий для обмена медицинской информацией
Телемедицина - использование компьютерных и телекоммуникационных технологий для обмена медицинской информацией