- Синдром Марфана и другие дисплазии соединительной ткани

Содержание
- 2. Содержание Соединительнотканная дисплазия Этиопатология Классификация Синдром Морфана Наследование Фенотип больного Симптомы синдрома Морфана Диагностика Лечение Литература
- 3. Соединительнотканная дисплазия – группа полиморфных в клиническом отношении патологических состояний, обусловленных наследственными или врожденными дефектами синтеза
- 4. Этиопатология ДСТ морфологически характеризуется изменениями коллагеновых, эластических фибрилл, гликопротеидов, протеогликанов и фибробластов, в основе которых лежат
- 5. Классификация Соединительнотканная дисплазия подразделяются на: дифференцированные недифференцированные. К числу дифференцированных дисплазий относятся заболевания с определенным, установленным
- 6. Синдром Морфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Это редкое заболевание с классическим
- 7. Наследуется по аутосомно-доминантному типу. В основе заболевания лежит нарушение синтеза одного из основных белков соединительной ткани
- 9. Больные синдромом Морфана, как правило, отличаются высоким ростом, относительно коротким туловищем с непропорционально длинными тонкими конечностями
- 10. Симптомы синдрома Марфана При синдроме Морфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной клетки (воронкообразная или
- 11. Признаки заболевания проявляются с момента рождения ребенка. В период новорожденности обнаруживаются скелетные аномалии в виде удлинения
- 12. Диагностика синдрома Марфана Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков
- 13. Диагностика синдрома Марфана За диагностические критерии синдрома Морфана берутся характерные изменения в различных системах и органах;
- 14. Согласно литературным данным у подавляющего большинства пациентов с ДСТ имеет место снижение уровня большинства макро- и
- 15. Дефицит меди приводит к нарушению коллагена и эластина, что способствует формированию аномалий развития сердечно-сосудистой системы и
- 17. Скачать презентацию

Содержание
Соединительнотканная дисплазия
Этиопатология
Классификация
Синдром Морфана
Наследование
Фенотип больного
Симптомы синдрома Морфана
Диагностика
Лечение
Литература
Содержание
Соединительнотканная дисплазия
Этиопатология
Классификация
Синдром Морфана
Наследование
Фенотип больного
Симптомы синдрома Морфана
Диагностика
Лечение
Литература

Соединительнотканная дисплазия – группа полиморфных в клиническом отношении патологических состояний, обусловленных наследственными
Соединительнотканная дисплазия – группа полиморфных в клиническом отношении патологических состояний, обусловленных наследственными

Этиопатология
ДСТ морфологически характеризуется изменениями коллагеновых, эластических фибрилл, гликопротеидов, протеогликанов и фибробластов,
Этиопатология
ДСТ морфологически характеризуется изменениями коллагеновых, эластических фибрилл, гликопротеидов, протеогликанов и фибробластов,

Классификация
Соединительнотканная дисплазия подразделяются на:
дифференцированные
недифференцированные.
К числу дифференцированных дисплазий относятся заболевания
Классификация
Соединительнотканная дисплазия подразделяются на:
дифференцированные
недифференцированные.
К числу дифференцированных дисплазий относятся заболевания

Синдром Морфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Это редкое
Синдром Морфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Это редкое

Наследуется по аутосомно-доминантному типу.
В основе заболевания лежит нарушение синтеза одного из
Наследуется по аутосомно-доминантному типу.
В основе заболевания лежит нарушение синтеза одного из
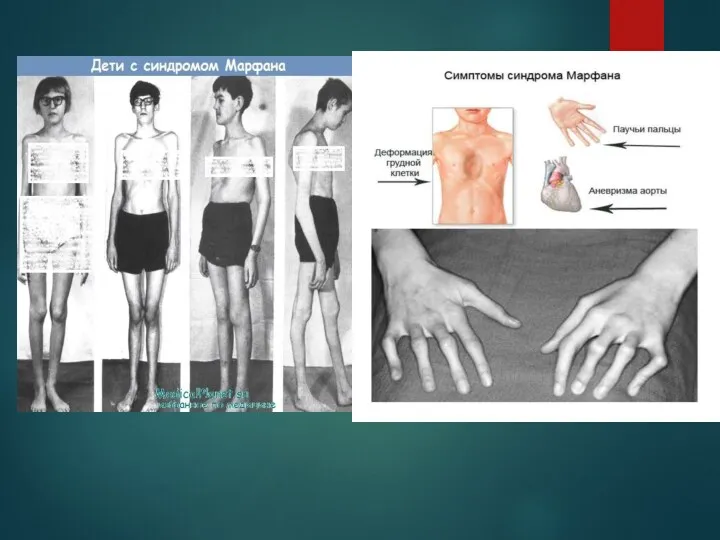

Больные синдромом Морфана, как правило, отличаются высоким ростом, относительно коротким туловищем
Больные синдромом Морфана, как правило, отличаются высоким ростом, относительно коротким туловищем

Симптомы синдрома Марфана
При синдроме Морфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной
Симптомы синдрома Марфана
При синдроме Морфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной

Признаки заболевания проявляются с момента рождения ребенка. В период новорожденности обнаруживаются
Признаки заболевания проявляются с момента рождения ребенка. В период новорожденности обнаруживаются

Диагностика синдрома Марфана
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у
Диагностика синдрома Марфана
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у

Диагностика синдрома Марфана
За диагностические критерии синдрома Морфана берутся характерные изменения в
Диагностика синдрома Марфана
За диагностические критерии синдрома Морфана берутся характерные изменения в

Согласно литературным данным у подавляющего большинства пациентов с ДСТ имеет место
Согласно литературным данным у подавляющего большинства пациентов с ДСТ имеет место

Дефицит меди приводит к нарушению коллагена и эластина, что способствует формированию
Дефицит меди приводит к нарушению коллагена и эластина, что способствует формированию
 Мужское бесплодие
Мужское бесплодие Cовременные представления о смазанном слое и гибридной зоне. Адгезия
Cовременные представления о смазанном слое и гибридной зоне. Адгезия Интенсивная терапия при гиповолемическом и геморрагическом шоке у детей
Интенсивная терапия при гиповолемическом и геморрагическом шоке у детей Лабораторные, инструментальные и функциональные методы исследования системы органов дыхания
Лабораторные, инструментальные и функциональные методы исследования системы органов дыхания Гранулематоз Вегенера М 31.3
Гранулематоз Вегенера М 31.3 Здорове харчування. Поживні речовини
Здорове харчування. Поживні речовини Морфологические особенности эритроцитов при анемиях. Картина крови при железодифицитной анемии
Морфологические особенности эритроцитов при анемиях. Картина крови при железодифицитной анемии Вирусные гипатиты
Вирусные гипатиты Лабораторная диагностика сифилиса и инфекций передаваемых половым путем
Лабораторная диагностика сифилиса и инфекций передаваемых половым путем Организация работы медицинской сестры процедурного кабинета
Организация работы медицинской сестры процедурного кабинета Методы исследования больных с заболеваниями органов кроветворения
Методы исследования больных с заболеваниями органов кроветворения Ерте кезеңдегі токсикоз
Ерте кезеңдегі токсикоз Ситуационные задачи. Роль ТГЗ в системе ЛЭО войск
Ситуационные задачи. Роль ТГЗ в системе ЛЭО войск Повреждения органов брюшной полости
Повреждения органов брюшной полости Синдром Патау
Синдром Патау Шат аралық. Аналық және аталық шатаралықтың құрылысының ерекшеліктері
Шат аралық. Аналық және аталық шатаралықтың құрылысының ерекшеліктері Дегенеративно -диструктивные изменения позвоночника
Дегенеративно -диструктивные изменения позвоночника Жергілікті иммунитет. Кілегейлі қабықпен, өкпемен,терімен ассоцияланған лимфоидты тіндер
Жергілікті иммунитет. Кілегейлі қабықпен, өкпемен,терімен ассоцияланған лимфоидты тіндер Острая и хроническая недостаточность кровообращения
Острая и хроническая недостаточность кровообращения Особенности двигательной сферы у лиц с ДЦП
Особенности двигательной сферы у лиц с ДЦП Роль гормонов в обмене веществ, росте и развитии организма
Роль гормонов в обмене веществ, росте и развитии организма Патология МВС: семиотика и методы диагностики. Гломерулонефрит, пиелонефрит. Почечная недостаточность: острая, хроническая
Патология МВС: семиотика и методы диагностики. Гломерулонефрит, пиелонефрит. Почечная недостаточность: острая, хроническая Нематодозы. Аскаридоз
Нематодозы. Аскаридоз Расстройства ассоциативного процесса мышления
Расстройства ассоциативного процесса мышления Синдром покраснения глаза
Синдром покраснения глаза Исторические этапы развития отечественной хирургической стоматологии
Исторические этапы развития отечественной хирургической стоматологии Бабж – диарея
Бабж – диарея Мерез. Мерез ауруының өзектілігі
Мерез. Мерез ауруының өзектілігі