- Специфические пути обмена отдельных аминокислот

Содержание
- 2. Обмен аминокислот: источники и пути использования
- 3. Общие пути катаболизма аминокислот в клетках Дезаминирование (отщепление аминогруппы от АК) Трансаминирование (переаминирование- перенос аминогруппы на
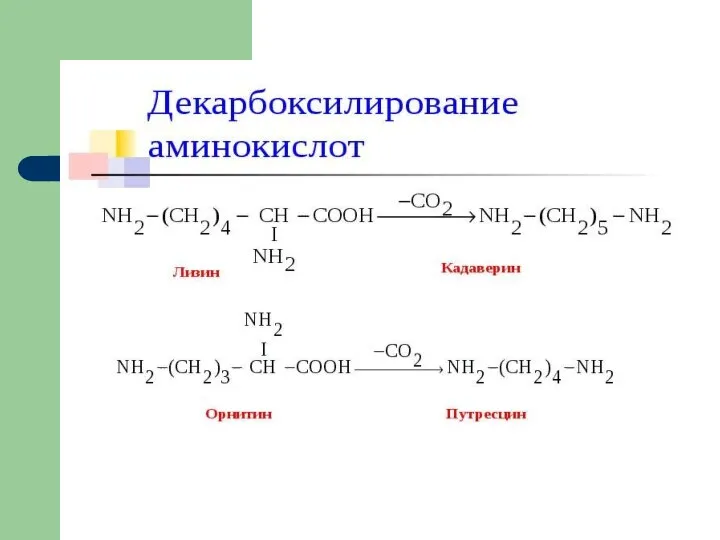
- 5. Судьба продуктов катаболизма (дезаминирования) аминокислот
- 6. ПУТИ ИСПОЛЬЗОВАНИЯ ДИКАРБОНОВЫХ АМИНОКИСЛОТ
- 7. Включение безазотистых остатков аминокислот в ЦТК
- 10. ЭФФЕКТЫ ГИСТАМИНА
- 11. Клинические проявления действия гистамина и антигистаминовых препаратов (блокаторов Н- рецепторов)
- 13. 90% серотонина синтезируется в жкт из триптофана, получаемого с пищей.
- 18. МЕЛАТОНИН- производное серотонина
- 21. ОБМЕН ФЕНИЛАЛАНИНА и ТИРОЗИНА
- 25. Роль дофамина в организме Как гормон: повышает артериальное давление, частоту и силу сердечных сокращений; расслабляет гладкую
- 27. Нарушение обмена дофамина наблюдается при шизофрении. ( гиперсекреция в височной доле, или недостаток в др структурах.)
- 29. ИНАКТИВАЦИЯ БА
- 30. Ингибиторы моноаминоксидазы ИМАО, MAOI — биологически активные вещества, способные ингибировать фермент моноаминоксидазу, содержащийся в нервных окончаниях,
- 35. Химизм реакций окисления фенилаланина в тирозин
- 37. ФЕНИЛКЕТОНУРИЯ - тяжелое наследственное заболевание, которое характеризуется главным образом поражением нервной системы. Фенилкетонурия (ФКУ) – развивается
- 38. Отсутствие в печени фермента фенилаланингидроксилазы препятствует нормальному превращению фенилаланина пищи в тирозин Поэтому фенилаланин используется лишь
- 39. ОБМЕН ФЕНИЛАЛАНИНА при ФКУ
- 40. ПРОЯВЛЕНИЯ ФКУ При заболевании нарушаются обменные процессы, особенно важные для развивающегося мозга ребенка. В крови и
- 41. В патогенезе ФКУ имеют значение следующие механизмы: Прямое токсическое действие на ЦНС фенилаланина и его производных
- 42. Варианты ФКУ Фенилкетонурия 1. Классическая фенилкетонурия (ФКУ) описана А.Folling.,1934г. Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена,
- 43. Варианты ФКУ Фенилкетонурия 2. Впервые атипичная ФКУ описана I.Smith, 1974г. Заболевание связано с дефицитом дигидроптеридинредуктазы. Нарушается
- 44. Варианты ФКУ Фенилкетонурия 3. Этот вариант болезни описал S. Kaufman в 1978 г. Заболевание связано с
- 45. Материнская фенилкетонурия. Заболевание развивается у детей женщин, страдающих ФКУ и не получающих диету в зрелом возрасте.
- 46. Клинические проявления ФКУ При рождении больные фенилкетонурией не отличаются от других новорожденных. Манифестация ФКУ происходит обычно
- 47. Клинические проявления ФКУ По мере прогрессирования болезни могут наблюдаться эпилептиформные приступы - развернутые судорожные и бессудорожные
- 48. Клинические проявления ФКУ Проявлениями болезни служат: вялость ребенка, отсутствие интереса к окружающему; повышенная раздражительность, беспокойство; срыгивание,
- 51. Клинико-лабораторная диагностика ФКУ В течение многих лет соответствующим диагностическим тестом служит реакция между фенилпировиноградной кислотой, которая
- 53. Лечение фенилкетонурии и прогноз Если ничего не предпринимать, фенилкетонурия приводит к развивитию олигофрении. Главным способом лечения
- 55. Очень важно! Кроме диетотерапии необходим постоянный медицинский контроль за умственным и физическим развитием ребенка. Применение диетотерапии
- 56. ФКУ- проблема социальная
- 58. Альбинизм - наследственно обусловленное нарушение синтеза пигментов (меланинов: эумеланинов и феомеланинов) - дефект тирозиназы. Цвет кожи,
- 60. Альбинизм Альбиносы чаще рождаются в семьях чернокожих. Известны случаи частичного альбинизма. Люди с такой болезнью имеют
- 62. ОБМЕН СЕРИН И ГЛИЦИНА Серин и глицин превращаются друг в друга. Роль реакции состоит в образовании
- 64. Образованный в реакции распада серина до глицина N5,N10-метилен-тетрагидрофолат (активная форма витамина В9) при участии фермента метилен-ТГФК-редуктазы
- 65. Обмен метионина Метионин присоединяет аденозильный остаток и превращается в активную форму метионина – S-аденозилметионин, участвующий во
- 66. S-аденозилметионин
- 68. Нарушение обмена метионина: дефект метиленфолатредуктазы или цистатионин-синтазы Гомоцистеин, растворенный в плазме, провоцирует свободнорадикальное окисление липидов в
- 69. метаболизм цистеина Цистеин является чрезвычайно важной аминокислотой в связи с тем, что это единственный источник органической
- 70. Пути использования цистеина
- 71. Синтез таурина
- 72. функции таурина является обязательным компонентом активных форм желчных кислот играет роль внутриклеточного антиоксиданта, играет роль тормозного
- 74. Скачать презентацию
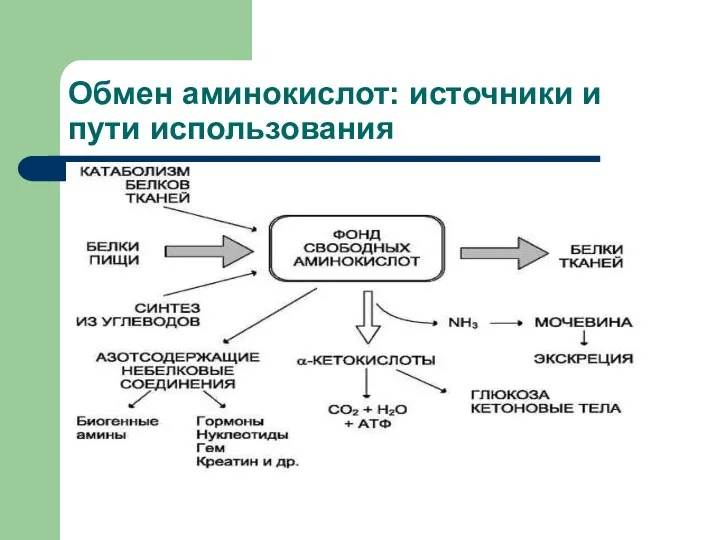
Обмен аминокислот: источники и пути использования
Обмен аминокислот: источники и пути использования

Общие пути катаболизма аминокислот в клетках
Дезаминирование (отщепление аминогруппы от АК)
Трансаминирование (переаминирование-
Общие пути катаболизма аминокислот в клетках
Дезаминирование (отщепление аминогруппы от АК)
Трансаминирование (переаминирование-

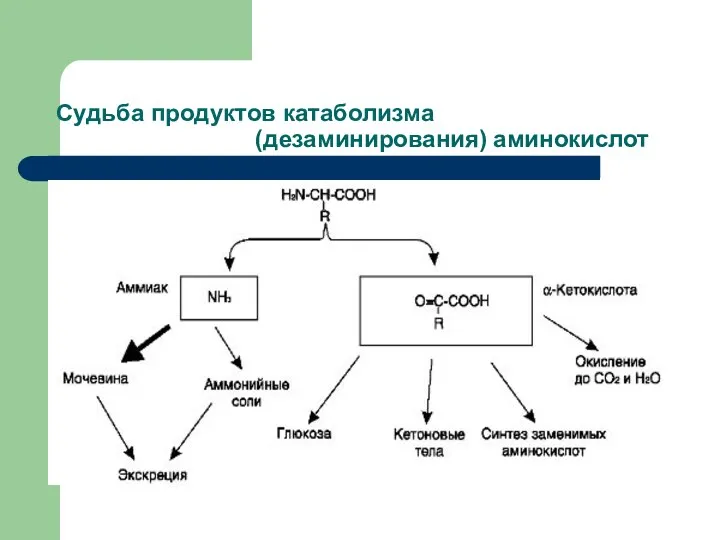
Судьба продуктов катаболизма (дезаминирования) аминокислот
Судьба продуктов катаболизма (дезаминирования) аминокислот
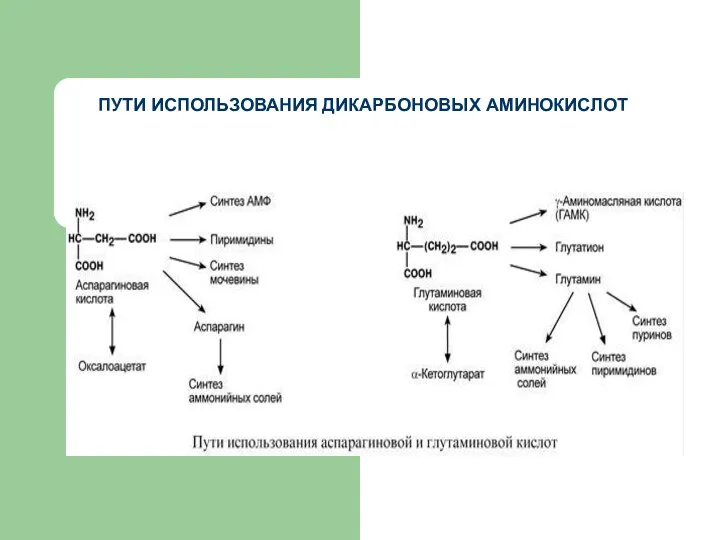
ПУТИ ИСПОЛЬЗОВАНИЯ ДИКАРБОНОВЫХ АМИНОКИСЛОТ
ПУТИ ИСПОЛЬЗОВАНИЯ ДИКАРБОНОВЫХ АМИНОКИСЛОТ
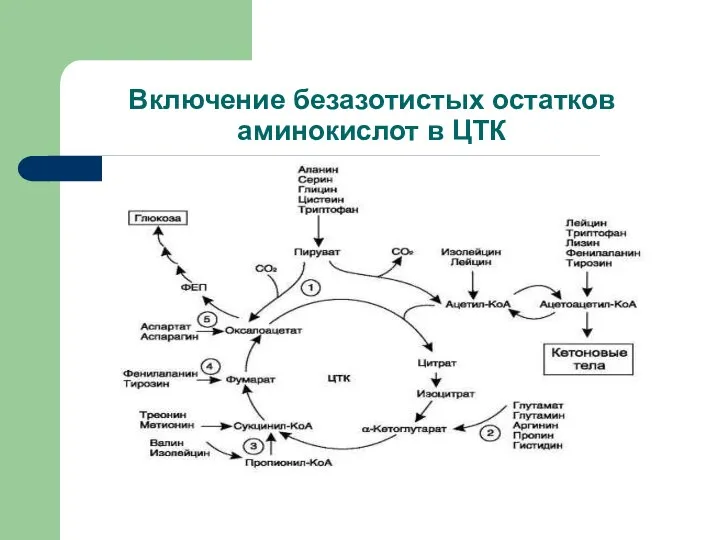
Включение безазотистых остатков аминокислот в ЦТК
Включение безазотистых остатков аминокислот в ЦТК


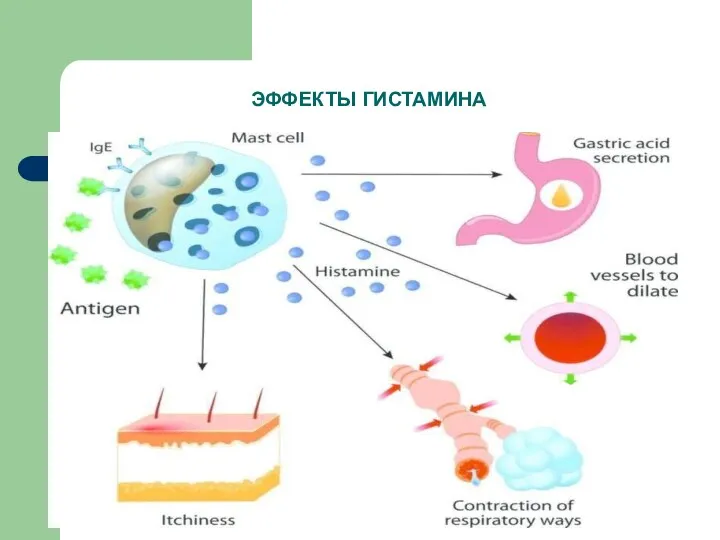
ЭФФЕКТЫ ГИСТАМИНА
ЭФФЕКТЫ ГИСТАМИНА
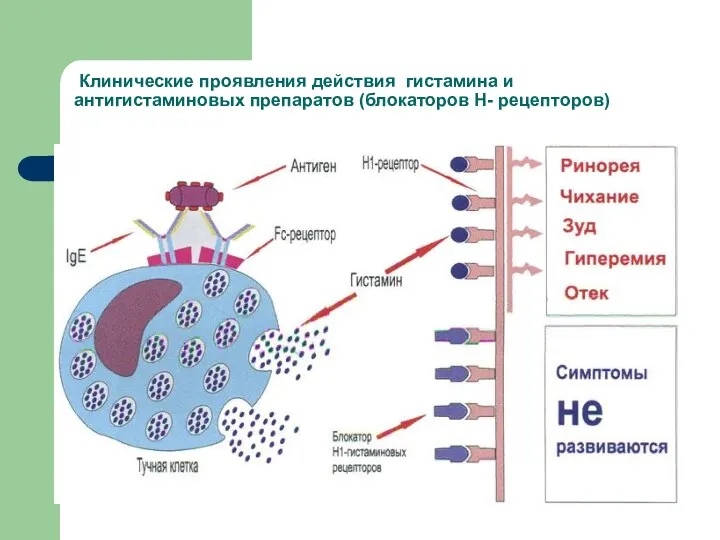
Клинические проявления действия гистамина и антигистаминовых препаратов (блокаторов Н- рецепторов)
Клинические проявления действия гистамина и антигистаминовых препаратов (блокаторов Н- рецепторов)

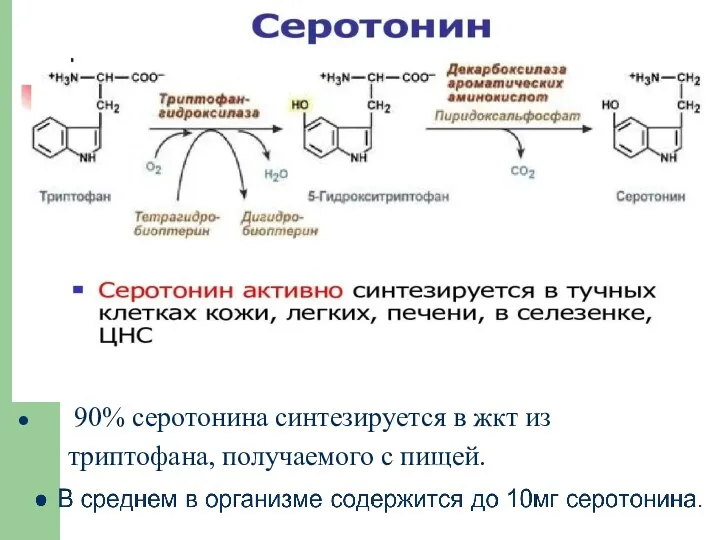
90% серотонина синтезируется в жкт из
триптофана, получаемого с пищей.
90% серотонина синтезируется в жкт из
триптофана, получаемого с пищей.




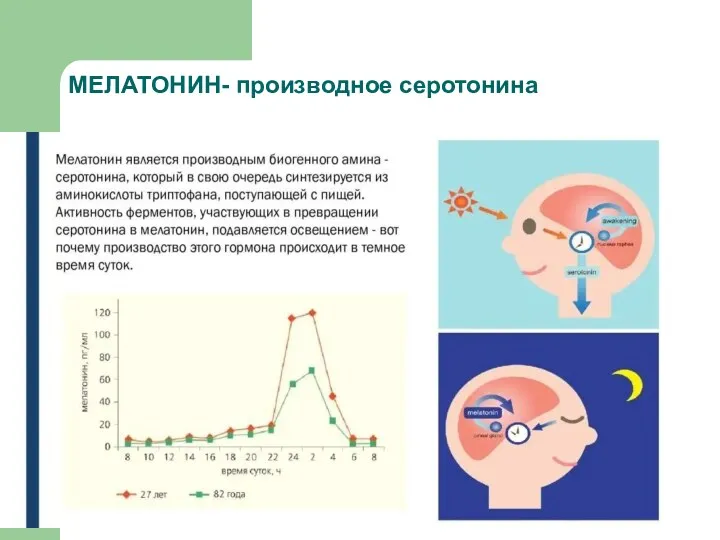
МЕЛАТОНИН- производное серотонина
МЕЛАТОНИН- производное серотонина


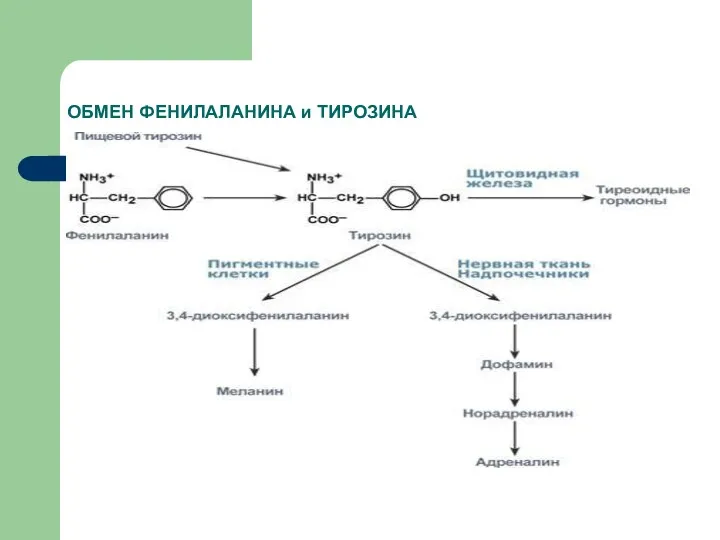
ОБМЕН ФЕНИЛАЛАНИНА и ТИРОЗИНА
ОБМЕН ФЕНИЛАЛАНИНА и ТИРОЗИНА
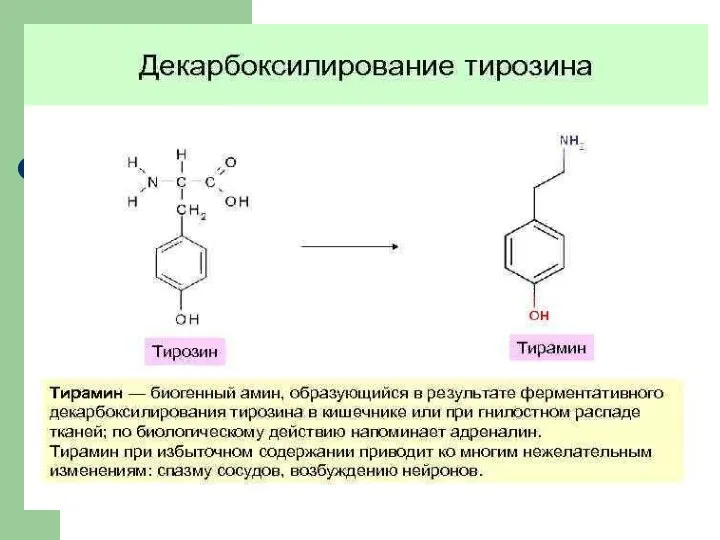

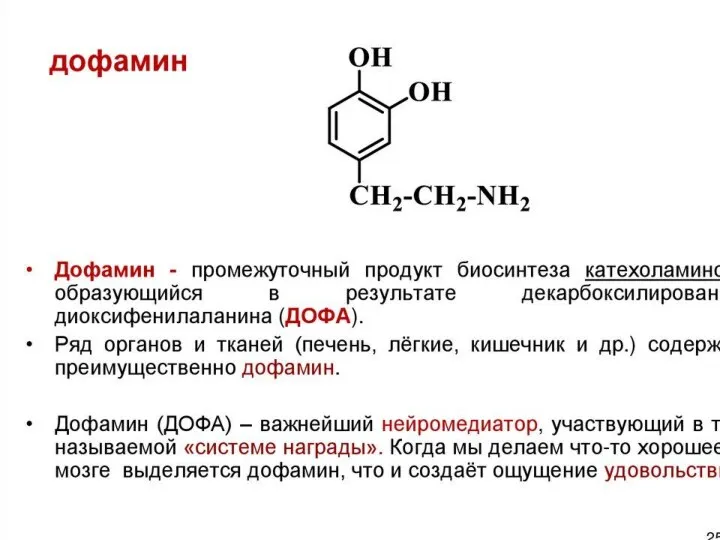

Роль дофамина в организме
Как гормон:
повышает артериальное давление, частоту и силу сердечных
Роль дофамина в организме
Как гормон:
повышает артериальное давление, частоту и силу сердечных


Нарушение обмена дофамина наблюдается при шизофрении. ( гиперсекреция в височной доле,


ИНАКТИВАЦИЯ БА
ИНАКТИВАЦИЯ БА

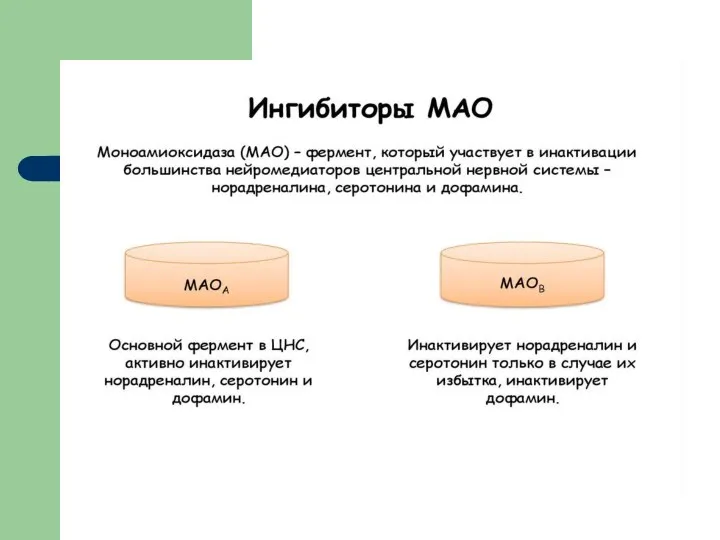
Ингибиторы моноаминоксидазы
ИМАО, MAOI — биологически активные вещества, способные ингибировать фермент
Ингибиторы моноаминоксидазы
ИМАО, MAOI — биологически активные вещества, способные ингибировать фермент


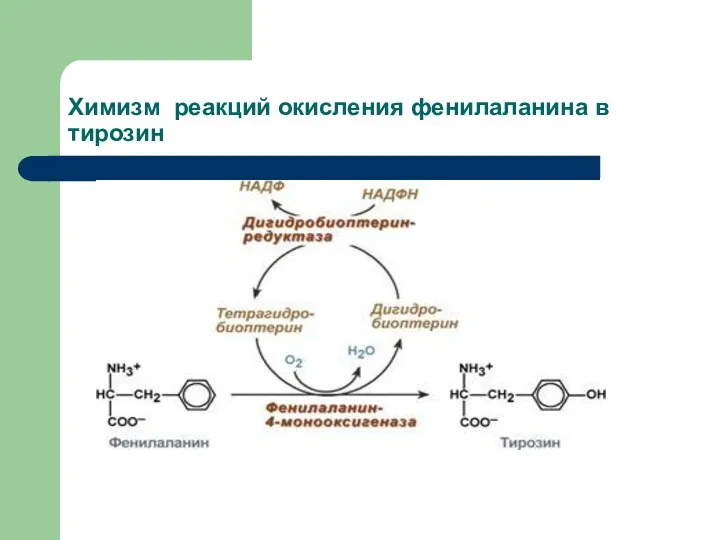
Химизм реакций окисления фенилаланина в тирозин
Химизм реакций окисления фенилаланина в тирозин


ФЕНИЛКЕТОНУРИЯ - тяжелое наследственное заболевание, которое характеризуется главным образом поражением нервной
ФЕНИЛКЕТОНУРИЯ - тяжелое наследственное заболевание, которое характеризуется главным образом поражением нервной

Отсутствие в печени фермента фенилаланингидроксилазы препятствует нормальному превращению фенилаланина пищи в
Отсутствие в печени фермента фенилаланингидроксилазы препятствует нормальному превращению фенилаланина пищи в
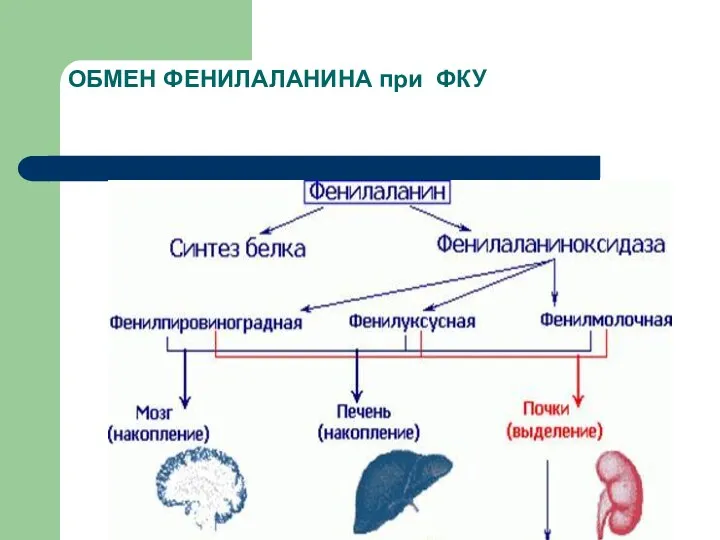
ОБМЕН ФЕНИЛАЛАНИНА при ФКУ
ОБМЕН ФЕНИЛАЛАНИНА при ФКУ

ПРОЯВЛЕНИЯ ФКУ
При заболевании нарушаются обменные процессы, особенно важные для развивающегося мозга
ПРОЯВЛЕНИЯ ФКУ
При заболевании нарушаются обменные процессы, особенно важные для развивающегося мозга

В патогенезе ФКУ имеют значение следующие механизмы:
Прямое токсическое действие на
В патогенезе ФКУ имеют значение следующие механизмы:
Прямое токсическое действие на

Варианты ФКУ
Фенилкетонурия 1.
Классическая фенилкетонурия (ФКУ) описана А.Folling.,1934г.
Заболевание
Варианты ФКУ
Фенилкетонурия 1.
Классическая фенилкетонурия (ФКУ) описана А.Folling.,1934г.
Заболевание

Варианты ФКУ
Фенилкетонурия 2.
Впервые атипичная ФКУ описана I.Smith, 1974г.
Заболевание связано
Варианты ФКУ
Фенилкетонурия 2.
Впервые атипичная ФКУ описана I.Smith, 1974г.
Заболевание связано

Варианты ФКУ
Фенилкетонурия 3. Этот вариант болезни описал S. Kaufman в 1978
Варианты ФКУ
Фенилкетонурия 3. Этот вариант болезни описал S. Kaufman в 1978

Материнская фенилкетонурия.
Заболевание развивается у детей женщин, страдающих ФКУ и не получающих
Материнская фенилкетонурия.
Заболевание развивается у детей женщин, страдающих ФКУ и не получающих

Клинические проявления ФКУ
При рождении больные фенилкетонурией не отличаются от других новорожденных.
Клинические проявления ФКУ
При рождении больные фенилкетонурией не отличаются от других новорожденных.

Клинические проявления ФКУ
По мере прогрессирования болезни могут наблюдаться эпилептиформные приступы -
Клинические проявления ФКУ
По мере прогрессирования болезни могут наблюдаться эпилептиформные приступы -

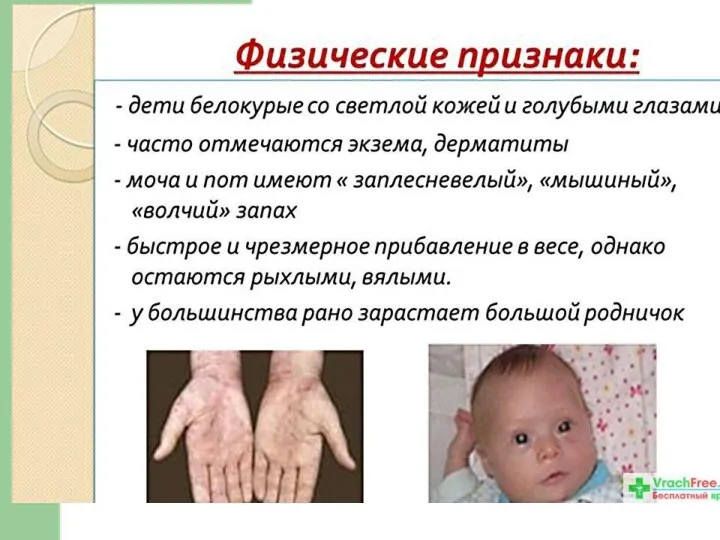
Клинические проявления ФКУ
Проявлениями болезни служат: вялость ребенка, отсутствие интереса к окружающему;
Клинические проявления ФКУ
Проявлениями болезни служат: вялость ребенка, отсутствие интереса к окружающему;


Клинико-лабораторная диагностика ФКУ
В течение многих лет соответствующим диагностическим тестом служит реакция
Клинико-лабораторная диагностика ФКУ
В течение многих лет соответствующим диагностическим тестом служит реакция


Лечение фенилкетонурии и прогноз
Если ничего не предпринимать, фенилкетонурия приводит к развивитию
Лечение фенилкетонурии и прогноз
Если ничего не предпринимать, фенилкетонурия приводит к развивитию


Очень важно!
Кроме диетотерапии необходим постоянный медицинский контроль за умственным и
Очень важно!
Кроме диетотерапии необходим постоянный медицинский контроль за умственным и

ФКУ- проблема социальная
ФКУ- проблема социальная


Альбинизм - наследственно обусловленное нарушение синтеза пигментов (меланинов: эумеланинов и феомеланинов)
Альбинизм - наследственно обусловленное нарушение синтеза пигментов (меланинов: эумеланинов и феомеланинов)


Альбинизм
Альбиносы чаще
рождаются в семьях чернокожих. Известны случаи частичного альбинизма. Люди
Альбинизм
Альбиносы чаще
рождаются в семьях чернокожих. Известны случаи частичного альбинизма. Люди

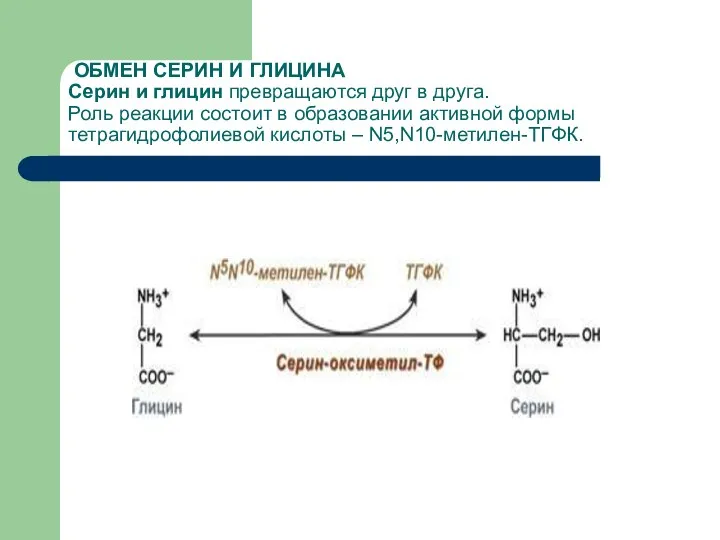
ОБМЕН СЕРИН И ГЛИЦИНА
Серин и глицин превращаются друг в друга.
Роль
ОБМЕН СЕРИН И ГЛИЦИНА Серин и глицин превращаются друг в друга. Роль


Образованный в реакции распада серина до глицина N5,N10-метилен-тетрагидрофолат (активная форма витамина В9)
Образованный в реакции распада серина до глицина N5,N10-метилен-тетрагидрофолат (активная форма витамина В9)

Обмен метионина
Метионин присоединяет аденозильный остаток и превращается в активную форму метионина
Обмен метионина
Метионин присоединяет аденозильный остаток и превращается в активную форму метионина
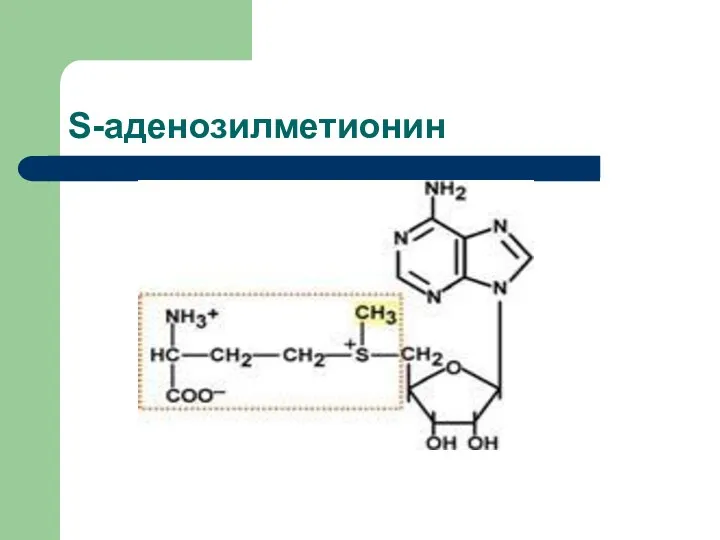
S-аденозилметионин
S-аденозилметионин
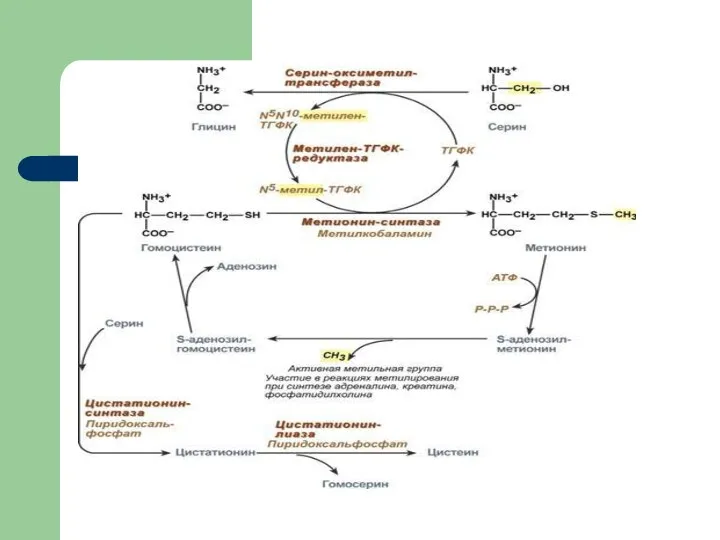

Нарушение обмена метионина:
дефект метиленфолатредуктазы или цистатионин-синтазы
Гомоцистеин, растворенный в
Нарушение обмена метионина:
дефект метиленфолатредуктазы или цистатионин-синтазы
Гомоцистеин, растворенный в

метаболизм цистеина
Цистеин является чрезвычайно важной аминокислотой в связи с тем,
метаболизм цистеина
Цистеин является чрезвычайно важной аминокислотой в связи с тем,
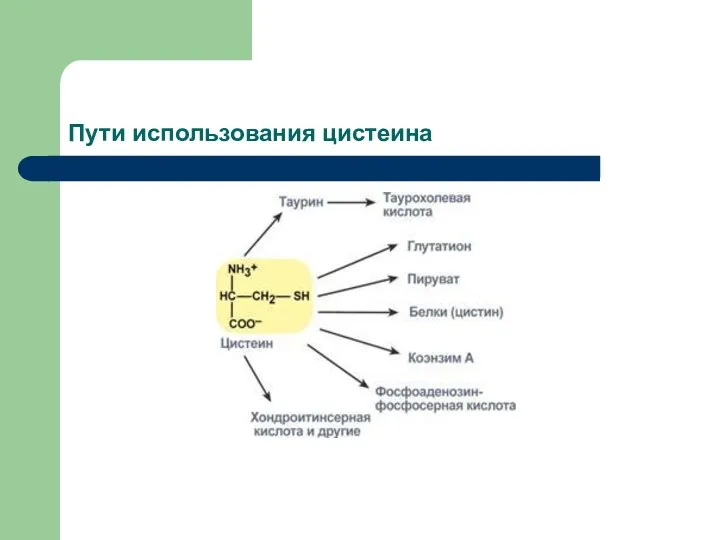
Пути использования цистеина
Пути использования цистеина
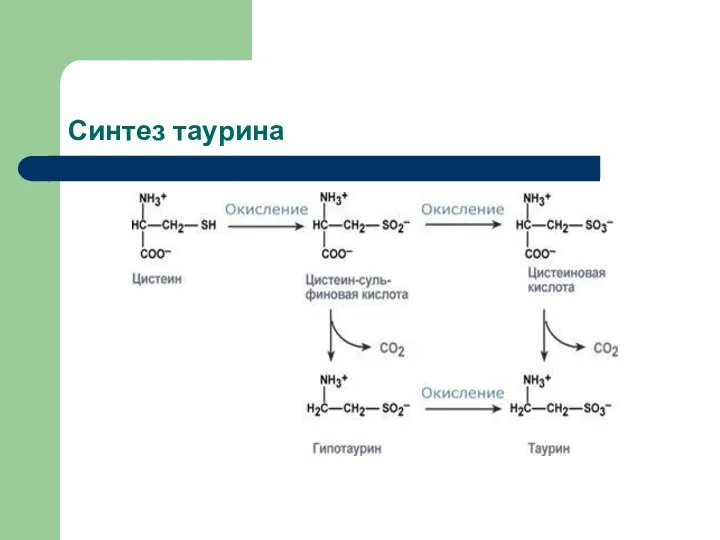
Синтез таурина
Синтез таурина

функции таурина
является обязательным компонентом активных форм желчных кислот
играет роль внутриклеточного антиоксиданта,
играет
функции таурина
является обязательным компонентом активных форм желчных кислот
играет роль внутриклеточного антиоксиданта,
играет
 Державна санітарно-епідеміологічна експертиза, як елемент соціально-гігієнічного моніторингу. Основні положення та організація
Державна санітарно-епідеміологічна експертиза, як елемент соціально-гігієнічного моніторингу. Основні положення та організація Операции на органах шеи
Операции на органах шеи Физиология паращитовидных желёз
Физиология паращитовидных желёз Повреждения и заболевания мочеполовых органов
Повреждения и заболевания мочеполовых органов Хирург Н.Н. Бурденко
Хирург Н.Н. Бурденко Арбовирусты инфекциялар. Кенелік энцефалит вирусы
Арбовирусты инфекциялар. Кенелік энцефалит вирусы Венозный возврат (ВВ) – приток венозной крови к сердцу
Венозный возврат (ВВ) – приток венозной крови к сердцу Шум и вибрация
Шум и вибрация Алкогольный цирроз
Алкогольный цирроз Возрастные особенности системы крови и иммунитета
Возрастные особенности системы крови и иммунитета Неврозы
Неврозы Противоаритмические лекарственные средства
Противоаритмические лекарственные средства Здоровье на работе. Что должен знать о ВИЧ/СПИДе каждый?
Здоровье на работе. Что должен знать о ВИЧ/СПИДе каждый? Гигиена аптечных заведений
Гигиена аптечных заведений Гиперчувствительность. Иммунодефициты. Аутоиммунные процессы
Гиперчувствительность. Иммунодефициты. Аутоиммунные процессы Послеродовые депрессии
Послеродовые депрессии Аллергия. Стоматология
Аллергия. Стоматология 84-я Всероссийская научная конференция студентов и молодых ученых. Отчет. Секция: Общая хирургия
84-я Всероссийская научная конференция студентов и молодых ученых. Отчет. Секция: Общая хирургия Клинико-экономические исследования
Клинико-экономические исследования Химиотерапевтические лекарственные препараты, макролиды и азалиды
Химиотерапевтические лекарственные препараты, макролиды и азалиды Пороки сердца
Пороки сердца Асқорыту жолдарының қатерлі және қатерсіз ісіктері
Асқорыту жолдарының қатерлі және қатерсіз ісіктері Мировые демографические показатели рождаемость, смертность в развитых и развивающихся странах. Демографическая ситуация в Росси
Мировые демографические показатели рождаемость, смертность в развитых и развивающихся странах. Демографическая ситуация в Росси Классификация геморрагического васкулита
Классификация геморрагического васкулита Белки
Белки ЦМК СД в акушерстве и гинекологии ,
ЦМК СД в акушерстве и гинекологии , Medical Education in Japan
Medical Education in Japan Заболевания органов пищеварения у пожилых людей
Заболевания органов пищеварения у пожилых людей