Сравнительная характеристика процедур регистрации лекарственных препаратов в России, странах ЕС и США презентация
- Сравнительная характеристика процедур регистрации лекарственных препаратов в России, странах ЕС и США

Содержание
- 2. На сегодняшний день мы имеем многолетний опыт регистрации лекарственных средств для ветеринарного применения во всем мире.
- 3. Уполномоченный Регистрационный орган Закон или директива, регулирующая процедуру регистрации Основной экспертный орган/учреждение Российская Федерация Евросоюз Соединенные
- 4. Форма представления регистрационного досье Российская Федерация Евросоюз Соединенные штаты Америки Бумажная Бумажная, электронная При централизованной процедуре
- 5. Структура регистрационного досье Нет четкой структуры Условно можно выделить: - Административную часть (инструкция по применению, маркировка,
- 6. Виды и основные этапы регистрационной процедуры в Российской Федерации В процессе одноэтапной процедуры регистрации проводится экспертиза
- 7. Виды и основные этапы регистрационной процедуры в Евросоюзе 1. Централизованная процедура Процедура, при которой лекарственное средство
- 8. Виды и основные этапы регистрационной процедуры в Евросоюзе 2. Процедура «взаимного признания». Процедура взаимного признания другими
- 9. Виды и основные этапы регистрационной процедуры в Соединенных штатах Америки Процедура одобрения новых лекарственных препаратов для
- 10. Виды и основные этапы регистрационной процедуры в Соединенных штатах Америки 2. Процедура одобрения дженериков. После того,
- 11. Продолжительность процедуры регистрации Российская Федерация Евросоюз Соединенные штаты Америки До 160 рабочих дней (ранее 210 дней)
- 12. Срок действия первой регистрации и частота ее подтверждения. Продолжительность процедуры подтверждения регистрации Российская Федерация Евросоюз Соединенные
- 13. Особенности процедуры внесения изменений в регистрационную документацию Российская Федерация Евросоюз 1. В случае внесения изменений в
- 14. Особенности процедуры внесения изменений в регистрационную документацию Соединенные штаты Америки В ряде случаев, обозначенных как категория
- 15. Продолжительность процедуры внесения изменений в регистрационную документацию/регистрационную лицензию Российская Федерация Евросоюз Соединенные штаты Америки Продолжительность процедуры
- 16. Создание базового документа в Евразийской экономической комиссии Российская Федерация, Республика Беларусь, Республика Казахстан, Армения Единая процедура
- 18. Скачать презентацию
На сегодняшний день мы имеем многолетний опыт регистрации лекарственных средств для
На сегодняшний день мы имеем многолетний опыт регистрации лекарственных средств для
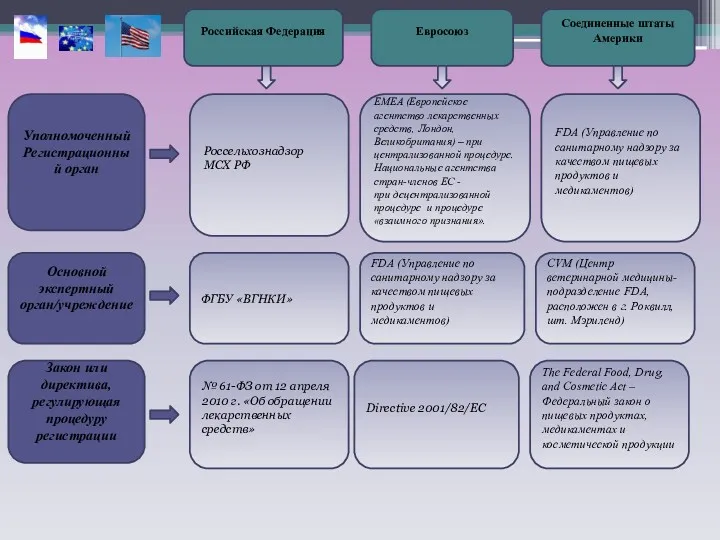
Уполномоченный Регистрационный орган
Закон или директива, регулирующая процедуру регистрации
Основной экспертный орган/учреждение
Российская Федерация
Евросоюз
Соединенные
Уполномоченный Регистрационный орган
Закон или директива, регулирующая процедуру регистрации
Основной экспертный орган/учреждение
Российская Федерация
Евросоюз
Соединенные
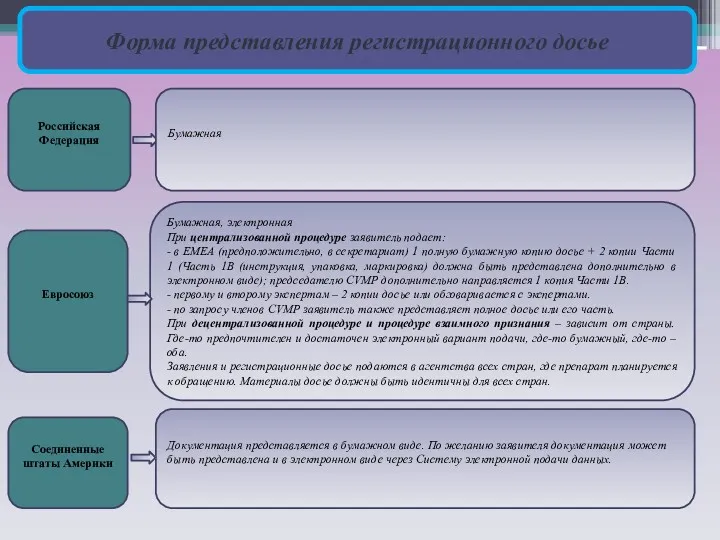
Форма представления регистрационного досье
Российская Федерация
Евросоюз
Соединенные штаты Америки
Бумажная
Бумажная, электронная
При централизованной
Форма представления регистрационного досье
Российская Федерация
Евросоюз
Соединенные штаты Америки
Бумажная
Бумажная, электронная
При централизованной

Структура регистрационного досье
Нет четкой структуры
Условно можно выделить:
- Административную часть (инструкция по
Структура регистрационного досье
Нет четкой структуры
Условно можно выделить:
- Административную часть (инструкция по
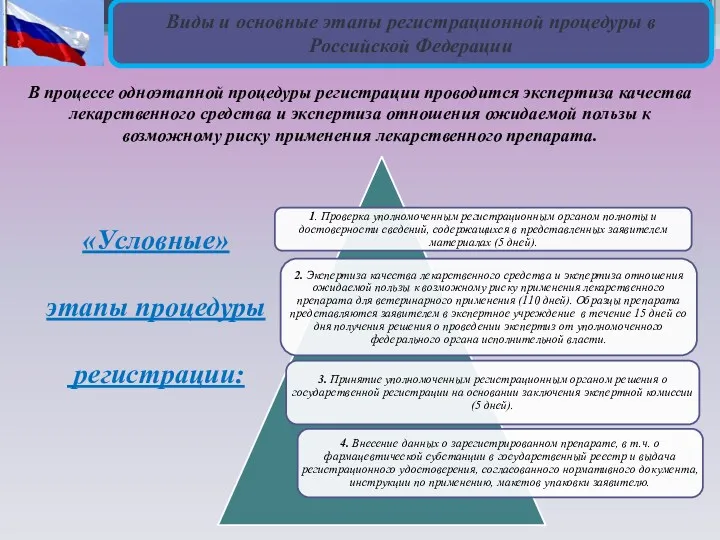
Виды и основные этапы регистрационной процедуры в Российской Федерации
В процессе одноэтапной
Виды и основные этапы регистрационной процедуры в Российской Федерации
В процессе одноэтапной

Виды и основные этапы регистрационной процедуры в Евросоюзе
1. Централизованная процедура
Процедура, при
Виды и основные этапы регистрационной процедуры в Евросоюзе
1. Централизованная процедура
Процедура, при

Виды и основные этапы регистрационной процедуры в Евросоюзе
2. Процедура «взаимного признания».
Процедура
Виды и основные этапы регистрационной процедуры в Евросоюзе
2. Процедура «взаимного признания».
Процедура

Виды и основные этапы регистрационной процедуры в Соединенных штатах Америки
Процедура одобрения
Виды и основные этапы регистрационной процедуры в Соединенных штатах Америки
Процедура одобрения

Виды и основные этапы регистрационной процедуры в Соединенных штатах Америки
2. Процедура
Виды и основные этапы регистрационной процедуры в Соединенных штатах Америки
2. Процедура
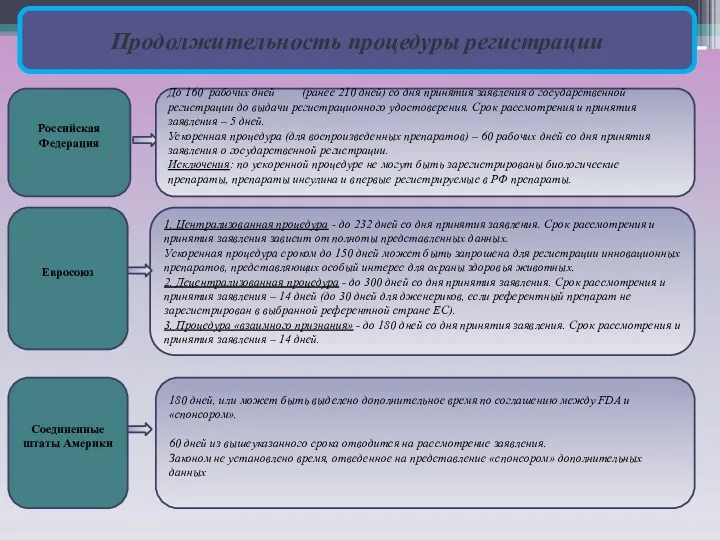
Продолжительность процедуры регистрации
Российская Федерация
Евросоюз
Соединенные штаты Америки
До 160 рабочих дней (ранее 210
Продолжительность процедуры регистрации
Российская Федерация
Евросоюз
Соединенные штаты Америки
До 160 рабочих дней (ранее 210
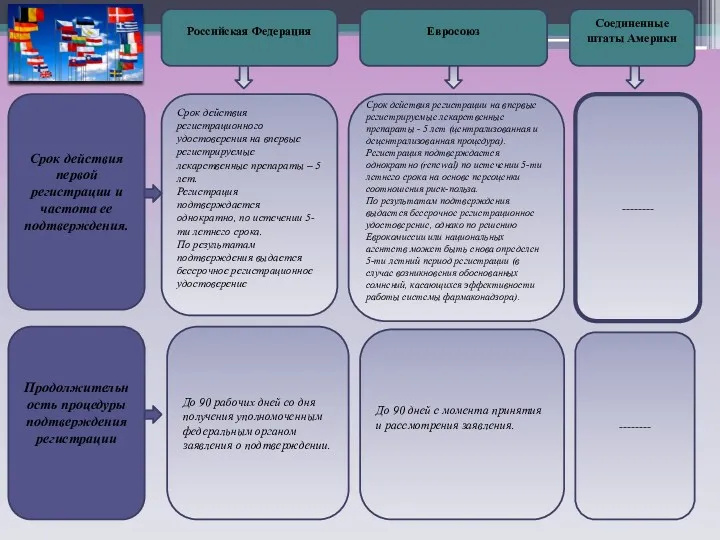
Срок действия первой регистрации и частота ее подтверждения.
Продолжительность процедуры подтверждения регистрации
Российская
Срок действия первой регистрации и частота ее подтверждения.
Продолжительность процедуры подтверждения регистрации
Российская

Особенности процедуры внесения изменений в регистрационную документацию
Российская Федерация
Евросоюз
1. В случае внесения
Особенности процедуры внесения изменений в регистрационную документацию
Российская Федерация
Евросоюз
1. В случае внесения

Особенности процедуры внесения изменений в регистрационную документацию
Соединенные штаты Америки
В ряде случаев,
Особенности процедуры внесения изменений в регистрационную документацию
Соединенные штаты Америки
В ряде случаев,
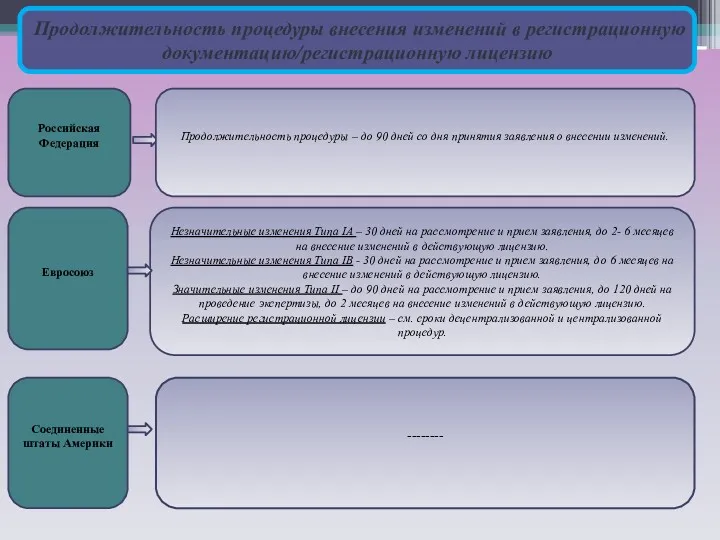
Продолжительность процедуры внесения изменений в регистрационную документацию/регистрационную лицензию
Российская Федерация
Евросоюз
Соединенные штаты Америки
Продолжительность
Продолжительность процедуры внесения изменений в регистрационную документацию/регистрационную лицензию
Российская Федерация
Евросоюз
Соединенные штаты Америки
Продолжительность
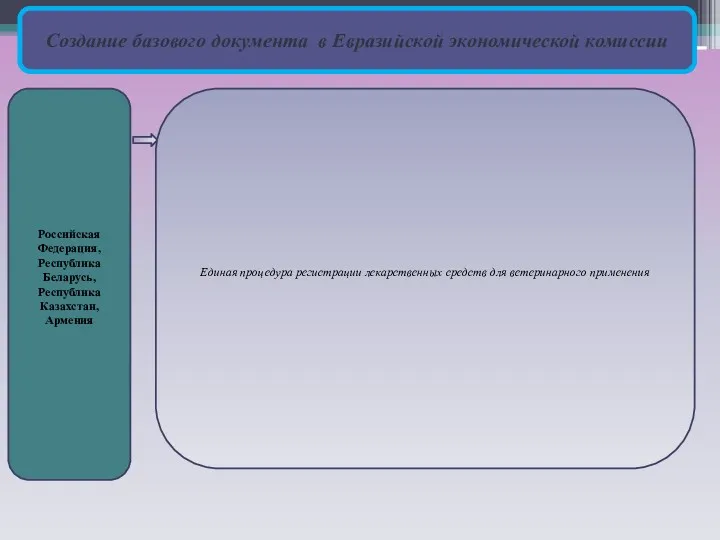
Создание базового документа в Евразийской экономической комиссии
Российская Федерация,
Республика Беларусь, Республика Казахстан,
Армения
Единая
Создание базового документа в Евразийской экономической комиссии
Российская Федерация,
Республика Беларусь, Республика Казахстан,
Армения
Единая
 Аномалии родовой деятельности
Аномалии родовой деятельности Лекция 2,3. Валеологический анализ факторов здоровья
Лекция 2,3. Валеологический анализ факторов здоровья Low-grade глиомы
Low-grade глиомы Мета та завдання стоматології
Мета та завдання стоматології Терминалды жағдай: дәрежелері, клиникасы, диагностикасы, науқас жағдайының ауырлығын бағалау критерилері
Терминалды жағдай: дәрежелері, клиникасы, диагностикасы, науқас жағдайының ауырлығын бағалау критерилері Сбор, хранение и транспортировка материала для микробиологических исследований
Сбор, хранение и транспортировка материала для микробиологических исследований Экстракорпоральные методы лечения критических состояний
Экстракорпоральные методы лечения критических состояний Қан топтары. Қан құю, тарихы және негізгі заңдылықтары. Құйылған қанның денеге әсері
Қан топтары. Қан құю, тарихы және негізгі заңдылықтары. Құйылған қанның денеге әсері Актуальные вопросы фибрилляции и трепетания предсердий
Актуальные вопросы фибрилляции и трепетания предсердий Нуклеопротеидтер алмасуының бұзылуы: подагра, несеп тас ауруы, несеп қышқыл, инфаркт
Нуклеопротеидтер алмасуының бұзылуы: подагра, несеп тас ауруы, несеп қышқыл, инфаркт Особо-опасные и актуальные для Ростовской области инфекции
Особо-опасные и актуальные для Ростовской области инфекции Культура общения медсестры с пациентом
Культура общения медсестры с пациентом Ситуационные задачи. Роль ТГЗ в системе ЛЭО войск
Ситуационные задачи. Роль ТГЗ в системе ЛЭО войск Характеристика психовегетативных расстройств у детей с артериальной гипертензией
Характеристика психовегетативных расстройств у детей с артериальной гипертензией Правила безопасного сексуального поведения
Правила безопасного сексуального поведения Распространенность факторов риска хронических неинфекционных заболеваний у населения Республики Коми с 2015 по 2017 год
Распространенность факторов риска хронических неинфекционных заболеваний у населения Республики Коми с 2015 по 2017 год Значение первой помощи и ухода за больными в системе медицинского образования
Значение первой помощи и ухода за больными в системе медицинского образования Лекарственные средства, производные конденсированных гетероциклических систем. (Тема 5)
Лекарственные средства, производные конденсированных гетероциклических систем. (Тема 5) Ожоговая болезнь, ее периоды (фазы). Организация лечебной помощи при ожогах на современном этапе. Ожоговый шок
Ожоговая болезнь, ее периоды (фазы). Организация лечебной помощи при ожогах на современном этапе. Ожоговый шок Basics of parasitic diseases in surgery
Basics of parasitic diseases in surgery Стоматологическое просвещение детей с ограниченными возможностями здоровья
Стоматологическое просвещение детей с ограниченными возможностями здоровья Денсаулы сатау жйесіндегі деректерге сараптама жасау. statistics пакетін қолдану
Денсаулы сатау жйесіндегі деректерге сараптама жасау. statistics пакетін қолдану Гипертензивные кризы, диагностика и лечение
Гипертензивные кризы, диагностика и лечение Side effects of chemotherapeutic drugs, cytostatics, hormonal medications
Side effects of chemotherapeutic drugs, cytostatics, hormonal medications Введение в нейрохирургию. Методы диагностики нейрохирургической патологии
Введение в нейрохирургию. Методы диагностики нейрохирургической патологии HELLP синдромы
HELLP синдромы Физиотерапевтические методы лечения заболеваний слизистой оболочки полости рта у детей
Физиотерапевтические методы лечения заболеваний слизистой оболочки полости рта у детей Ботулизм және тағамдық токсикоинфекция
Ботулизм және тағамдық токсикоинфекция