Врожденные нарушения метаболизма, вызывающие поражение ЖКТ у детей (гемохроматоз, тиразиноз, болезнь Вильсона-Коновалова) презентация
- Врожденные нарушения метаболизма, вызывающие поражение ЖКТ у детей (гемохроматоз, тиразиноз, болезнь Вильсона-Коновалова)

Содержание
- 2. Определение гемохроматоз тяжелое многосистемное заболевание, связанное с генетическими дефектами, определяющими повышение всасывания железа в желудочно-кишечном тракте,
- 3. Классификация HFE ( классическая форма)- классическая триада признаков, часто в сочетании симптомами поражения сердца и эндокринных
- 4. Патогенез НГ. Токсическое воздействие железа: усиление перекисного окисления липидов за счет катализирования железом свободнорадикальных реакции; усиление
- 7. Клинические проявления гемохроматоза Начальные симптомы гемохроматоза: Слабость Утомляемость Потеря веса Изменения окраски кожи (дымчатая) Боли в
- 8. Дифференциальную диагностику проводим при обнаружении: гепатомегалии неясного генеза; нелокализованных упорных болей в животе; сахарного диабета 2-го
- 9. Лечение диетотерапия: - запрет на введение железа - умеренное потребление мяса, исключить продукты с высоким содержанием
- 10. Фенилаланин – незаменимая аминокислота, так как в клетках животных не синтезируется ее бензольное кольцо, гликокетогенная. Тирозин
- 11. Тирозингидроксилаза (Fe2+) Адреналин Норадреналин Синтез катехоламинов (надпочечники, нервная ткань) О2 Н2О ДОФА-декарбоксилаза (В6) СО2 ДОФамингид- роксилаза
- 12. Фенилаланин Тирозин n-гидроксифенилпируват Гомогентизиновая кислота Фумарилацетоацетат Фумарат Ацетоацетат Катаболизм фенилаланина и тирозина в печени О2 Н2О
- 13. Белки (пищи и тканей) Фен Врожденные нарушения обмена ФЕН и ТИР Фенилпируват Фенилактат Фенилацетат Тир ДОФА
- 14. Нарушения катаболизма тирозина в печени приводит к тирозинемии и тирозинурии. Различают 3 типа тирозинемии: 1) Тирозинемия
- 15. 2) Тирозинемия типа 2 (Синдром Рихнера –Ханхорта). Причиной является дефект фермента тирозинаминотранс-феразы. Для заболевания характерны поражения
- 16. Болезнь Вильсона — Коновалова (гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона — Коновалова) — наследственное
- 17. Аутосомно-рецессивный тип наследования болезни Вильсона. 25 % вероятность рождения больного у родителей-гетерозигот У заболевания аутосомно-рецессивный тип
- 18. Патогенез Медь выполняет множество функций в организме. В основном она выступает в качестве кофактора для некоторых
- 19. Патогенетические стадии болезни Вильсона-Коновалов Начальный период накопления меди (преимущественно в печени). Распределение меди в печени и
- 20. Клиника Поражение печени протекает по типу хронического гепатита либо цирроза и клинически.Заболевание начинается остро, с развития
- 21. Отложение меди в десцеметовой мембране роговицы проявляется формированием колец Кайзера-Флейшера. В роговице отложение меди происходит почти
- 23. Диагностика Основой диагностики является картина болезни. Диагноз заболевания подтверждается: Наличием кольца Кайзера-ФлейшераНаличием кольца Кайзера-Флейшера или его
- 24. Инструментальные данные УЗИ и радиоизотопное сканирование печени: увеличение печени, селезенки, диффузные изменения. Биопсия печени: картина хронического
- 25. Синдром Алажиля — синдромальная форма патологии, включающая сочетание не менее трех из пяти основных признаков: хронический
- 26. ЭТИОЛОГИЯ Синдром Алажиля имеет аутосомно-доминантный тип наследования. Генный дефект связан с частичной делецией короткого плеча 20-й
- 27. ПАТОГЕНЕЗ В основе изменений печени при синдроме Алажиля лежит врожденная гипоплазия внутрипеченочных желчных протоков, степень выраженности
- 28. КЛИНИЧЕСКАЯ КАРТИНА Синдром холестаза появляется в период новорожденности, реже в течение первых месяцев жизни. Отмечается желтуха
- 29. ДИАГНОСТИКА Физикальное исследование Необходимо оценить цвет кожного покрова и склер, размеры печени и селезенки, цвет стула
- 30. КЛИНИЧЕСКАЯ КАРТИНА Возможны два варианта течения болезни. При легком варианте отмечают купирование клинических проявлений болезни к
- 31. Изменения глаз Наиболее типичное изменение — задний эмбриотоксон (малая аномалия развития в виде кольцевидного помутнения и
- 32. ЛЕЧЕНИЕ Патогенетическое. Цели лечения: коррекция осложнений длительно сохраняющегося холестаза. Немедикаментозное лечение Лечебное питание с повышенным содержанием
- 33. Дефицит А1АТ Аутосомно-рецессивное заболевание, вызываемое нарушением синтеза альфа 1-антитрипсина Пониженная активность А1АТ в крови и в
- 34. Альфа-1-антитрипсин – белок, который вырабатывается печенью. Он помогает организму в инактивации ферментов, при этом основная его
- 35. Синтез альфа-1-антитрипсина регулируется двумя копиями гена протеазного ингибитора серпина-1. Это так называемый кодоминантный ген, то есть
- 36. А1АТ Представитель семейства серпинов Серпины являются ингибиторами сериновых протеаз Основная функция – ингибирование эластазы Эластаза -
- 37. А1АТ: ген SERPINA1 (или Pi) 14q32.1 12,2 kbp 7 экзонов (4 кодирующих, 3 некодирующих), 6 интронов
- 38. Количество производимого альфа-1-антитрипсина и его активность зависят от типа унаследованной мутации. Несмотря на то что ген
- 39. А1АТ: белок (структура) 52 кДа 394 аминокислотных остатков, 3 гидрокарбонатные цепи RCL – reactive centre loop
- 40. А1АТ: белок (механизм ингибирования) (1) RCL ковалентно связывается с протеазой Конформационные изменения
- 41. А1АТ: белок (механизм ингибирования) (2) Протеаза атакует RCL RCL встраивается в бета-лист А, образуя четвёртый бета-лист
- 42. А1АТ: мутации Приводят к неправильному фолдингу, полимеризации, связыванию двух А1АТ друг с другом Наиболее важные мутации
- 43. Показания к опредению А1АТ: Если желтуха у новорождённого или малолетнего ребенка длится дольше 1-2 недель, при
- 44. Эмфизема Недостаток А1АТ (нормальное содержание – 1,5-3,5 г/л) => неконтролируемая активность протеаз, разрушение тканей лёгких (даже
- 45. Диагностирование 95% случаев – не диагностировано Содержание А1АТ в сыворотке или плазме крови (не определить гетерозиготы)
- 46. Лечение Лечение симптомов Регулярное введение А1АТ (внутривенные инфузии из донорной плазмы крови, аэрозоли) Использование других ингибиторов
- 47. Болезнь Байлера – это редкое наследственное (передающееся от родителей к детям) заболевание, в основе которого лежит
- 48. Симптомы болезнь байлера Заболевание проявляется синдромом холестаза. Для него характерен ряд симптомов. Главным симптомом холестаза является
- 49. Причины В основе синдрома и болезни Байлера лежит нарушение транспорта (переноса) желчных кислот (кислот, образующихся в
- 50. Диагностика Анализ анамнеза заболевания и жалоб пациента (когда (как давно) появились желтуха (пожелтение кожи и склер
- 51. Значения PGA колеблются от 0 до 12. Если PGA 9, то вероятность цирроза составляет 86%. Инструментальные
- 52. Лечение болезнь байлера Выделяют консервативное (безоперационное) и хирургическое лечение заболевания, а также общие рекомендации. К общим
- 53. Муковисцидоз - наследственная болезнь, характеризующаяся системным поражением экзокринных желез (внешней секреции) и проявляющаяся тяжелыми нарушениями функций
- 54. Из-за нарушения транспорта электролитов через мембрану клеток, которые выстилают протоки желез внешней секреции, выделяемый этими железами
- 55. I. Формы муковисцидоза • Смешанная (легочно-кишечная) с поражением желудочно-кишечного тракта и бронхолегочной системы (75—80%). • Легочная
- 56. симптомы со стороны дыхательной системы: хронический кашель, рецидивирующие пневмонии и ателектазы, перерастяжение легкого, барабанные палочки (своеобразная
- 57. При диагностике муковисцидоза учитываются данные клиники, истории развития ребенка и семьи, дополнительные исследования. Потовая проба Измерение
- 58. Недостаток витамина Е проявляется гемолитической анемией у новорожденных и неврологической симптоматикой у детей старшего возраста. Обструкция
- 59. Сахарный диабет выявляется у 20% взрослых пациентов с муковисцидозом. Фиброз печени, развивающийся в той или иной
- 61. Скачать презентацию

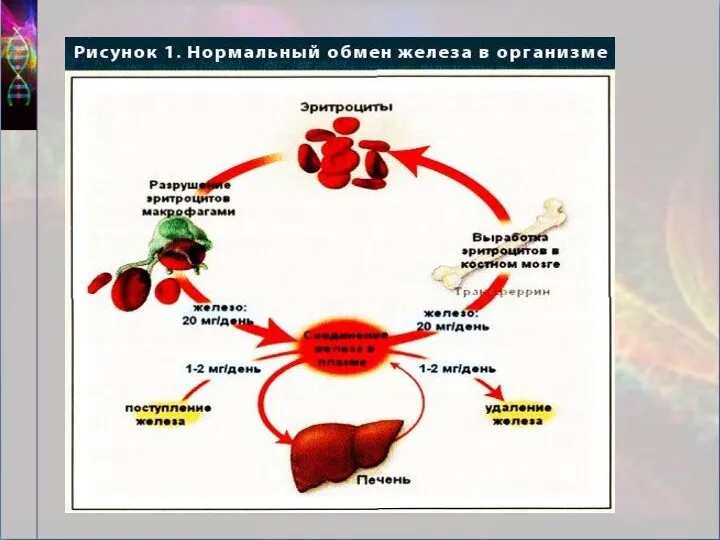
Определение гемохроматоз
тяжелое многосистемное заболевание, связанное с генетическими дефектами, определяющими повышение
Определение гемохроматоз
тяжелое многосистемное заболевание, связанное с генетическими дефектами, определяющими повышение

Классификация
HFE ( классическая форма)- классическая триада признаков, часто в сочетании
Классификация
HFE ( классическая форма)- классическая триада признаков, часто в сочетании

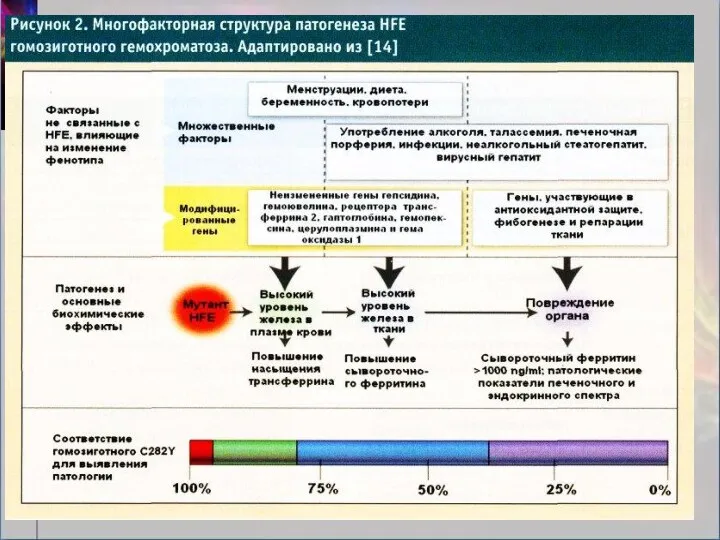
Патогенез НГ. Токсическое воздействие железа:
усиление перекисного окисления липидов за счет
Патогенез НГ. Токсическое воздействие железа:
усиление перекисного окисления липидов за счет
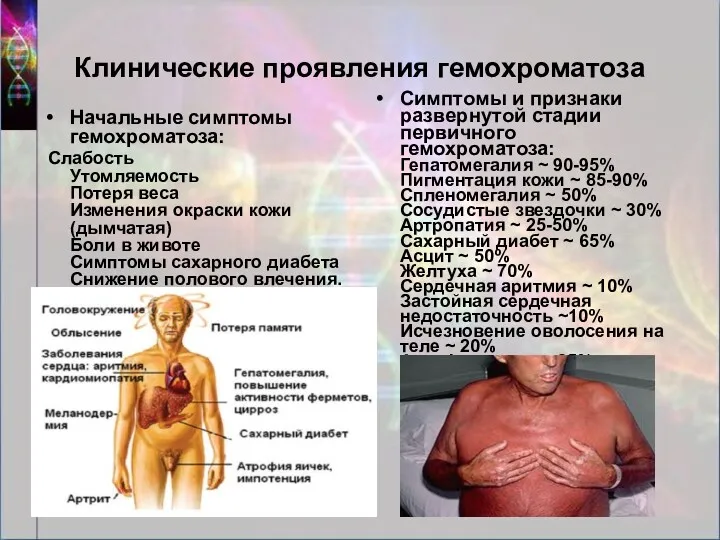
Клинические проявления гемохроматоза
Начальные симптомы гемохроматоза:
Слабость Утомляемость Потеря веса Изменения
Клинические проявления гемохроматоза
Начальные симптомы гемохроматоза:
Слабость Утомляемость Потеря веса Изменения

Дифференциальную диагностику проводим при обнаружении:
гепатомегалии неясного генеза;
нелокализованных упорных болей
Дифференциальную диагностику проводим при обнаружении:
гепатомегалии неясного генеза;
нелокализованных упорных болей

Лечение
диетотерапия:
- запрет на введение железа
- умеренное потребление мяса, исключить
Лечение
диетотерапия:
- запрет на введение железа
- умеренное потребление мяса, исключить

Фенилаланин – незаменимая аминокислота, так как в клетках животных не синтезируется
Фенилаланин – незаменимая аминокислота, так как в клетках животных не синтезируется
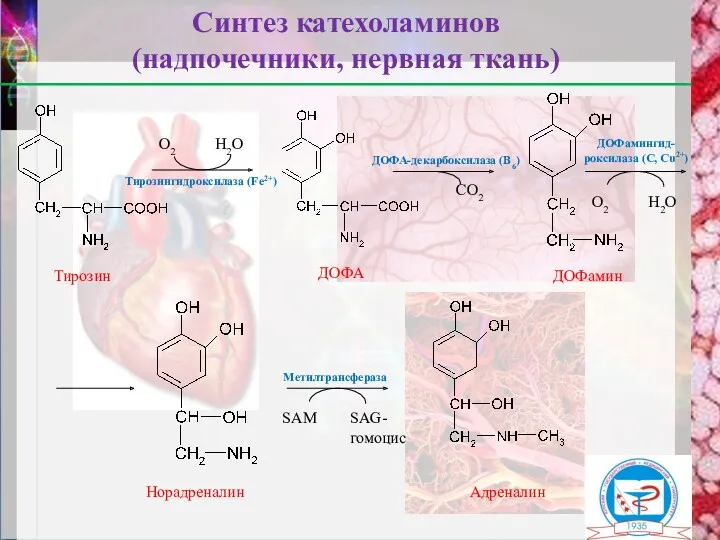
Тирозингидроксилаза (Fe2+)
Адреналин
Норадреналин
Синтез катехоламинов
(надпочечники, нервная ткань)
О2
Н2О
ДОФА-декарбоксилаза (В6)
СО2
ДОФамингид-
роксилаза (С, Сu2+)
О2
Н2О
Метилтрансфераза
SAM
SAG-гомоцис
Тирозингидроксилаза (Fe2+)
Адреналин
Норадреналин
Синтез катехоламинов
(надпочечники, нервная ткань)
О2
Н2О
ДОФА-декарбоксилаза (В6)
СО2
ДОФамингид-
роксилаза (С, Сu2+)
О2
Н2О
Метилтрансфераза
SAM
SAG-гомоцис
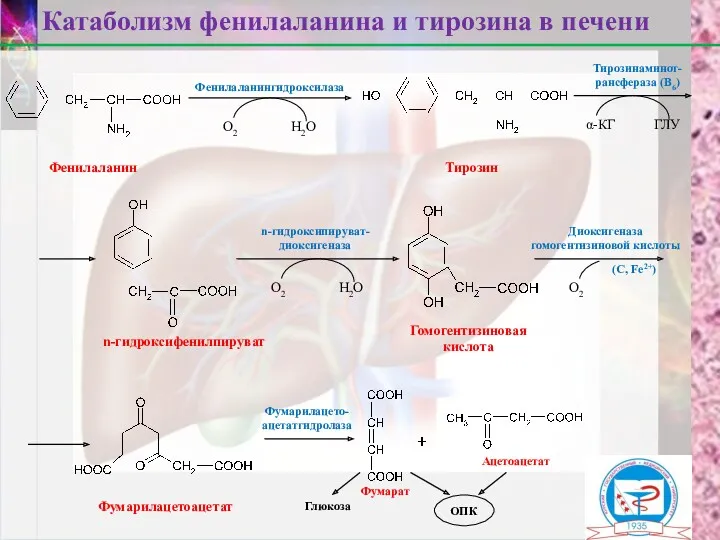
Фенилаланин
Тирозин
n-гидроксифенилпируват
Гомогентизиновая
кислота
Фумарилацетоацетат
Фумарат
Ацетоацетат
Катаболизм фенилаланина и тирозина в печени
О2
Н2О
Фенилаланингидроксилаза
α-КГ
ГЛУ
Тирозинаминот-
рансфераза (В6)
Фенилаланин
Тирозин
n-гидроксифенилпируват
Гомогентизиновая
кислота
Фумарилацетоацетат
Фумарат
Ацетоацетат
Катаболизм фенилаланина и тирозина в печени
О2
Н2О
Фенилаланингидроксилаза
α-КГ
ГЛУ
Тирозинаминот-
рансфераза (В6)
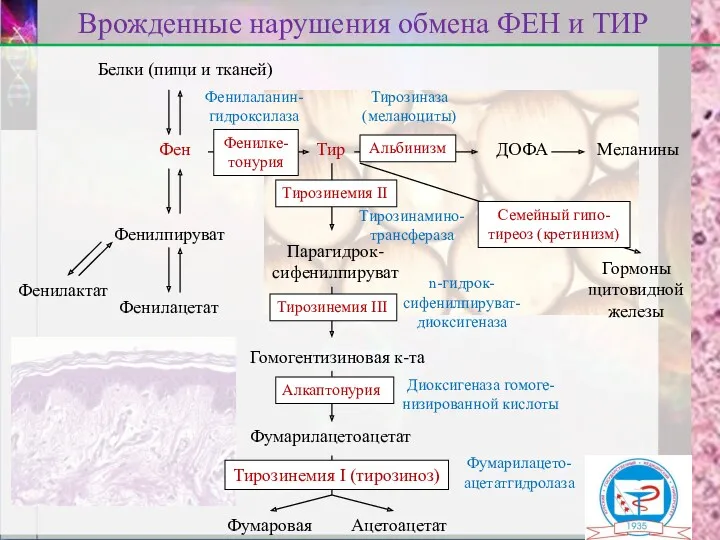
Белки (пищи и тканей)
Фен
Врожденные нарушения обмена ФЕН и ТИР
Фенилпируват
Фенилактат
Фенилацетат
Тир
ДОФА
Меланины
Гормоны
щитовидной
железы
Парагидрок-
сифенилпируват
n-гидрок-
сифенилпируват-
диоксигеназа
Фенилаланин-
гидроксилаза
Тирозиназа
(меланоциты)
Гомогентизиновая к-та
Диоксигеназа гомоге-
низированной
Белки (пищи и тканей)
Фен
Врожденные нарушения обмена ФЕН и ТИР
Фенилпируват
Фенилактат
Фенилацетат
Тир
ДОФА
Меланины
Гормоны
щитовидной
железы
Парагидрок-
сифенилпируват
n-гидрок-
сифенилпируват-
диоксигеназа
Фенилаланин-
гидроксилаза
Тирозиназа
(меланоциты)
Гомогентизиновая к-та
Диоксигеназа гомоге-
низированной
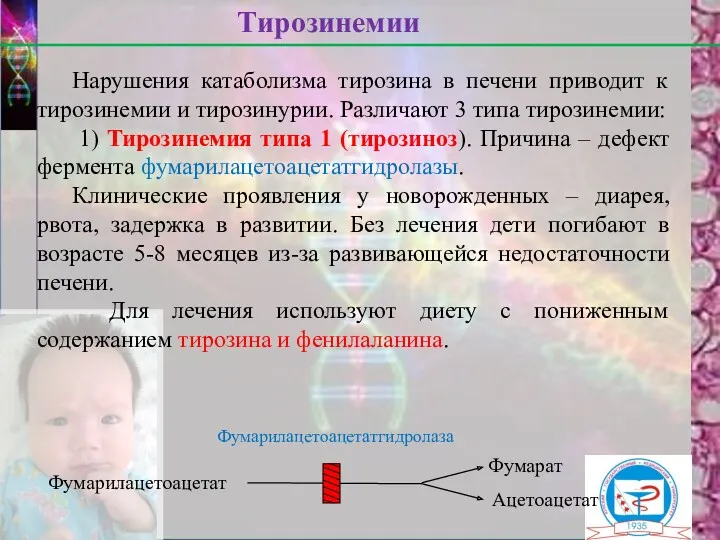
Нарушения катаболизма тирозина в печени приводит к тирозинемии и тирозинурии. Различают
Нарушения катаболизма тирозина в печени приводит к тирозинемии и тирозинурии. Различают
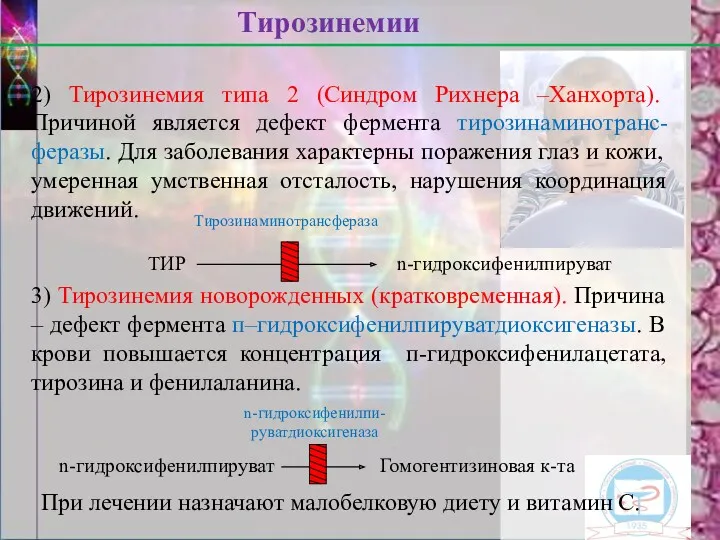
2) Тирозинемия типа 2 (Синдром Рихнера –Ханхорта). Причиной является дефект фермента

Болезнь Вильсона — Коновалова (гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона — Коновалова) —
Болезнь Вильсона — Коновалова (гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона — Коновалова) —

Аутосомно-рецессивный тип наследования болезни Вильсона. 25 % вероятность рождения больного у родителей-гетерозигот
Аутосомно-рецессивный тип наследования болезни Вильсона. 25 % вероятность рождения больного у родителей-гетерозигот

Патогенез
Медь выполняет множество функций в организме. В основном она выступает в
Патогенез
Медь выполняет множество функций в организме. В основном она выступает в

Патогенетические стадии болезни Вильсона-Коновалов
Начальный период накопления меди (преимущественно в печени).
Распределение меди
Патогенетические стадии болезни Вильсона-Коновалов
Начальный период накопления меди (преимущественно в печени).
Распределение меди

Клиника
Поражение печени протекает по типу хронического гепатита либо цирроза и клинически.Заболевание
Клиника
Поражение печени протекает по типу хронического гепатита либо цирроза и клинически.Заболевание

Отложение меди в десцеметовой мембране роговицы проявляется формированием колец Кайзера-Флейшера. В
Отложение меди в десцеметовой мембране роговицы проявляется формированием колец Кайзера-Флейшера. В
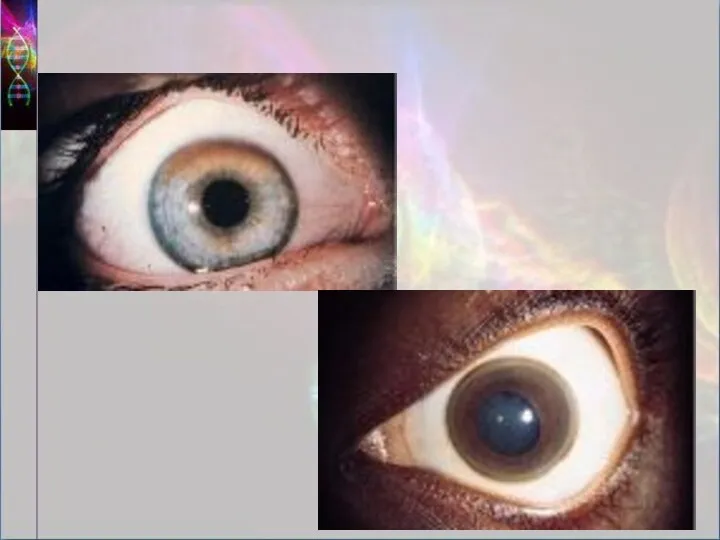

Диагностика
Основой диагностики является картина болезни. Диагноз заболевания подтверждается:
Наличием кольца Кайзера-ФлейшераНаличием кольца
Диагностика
Основой диагностики является картина болезни. Диагноз заболевания подтверждается:
Наличием кольца Кайзера-ФлейшераНаличием кольца

Инструментальные данные
УЗИ и радиоизотопное сканирование печени: увеличение печени, селезенки, диффузные
Инструментальные данные
УЗИ и радиоизотопное сканирование печени: увеличение печени, селезенки, диффузные

Синдром Алажиля — синдромальная форма патологии, включающая сочетание не менее трех
Синдром Алажиля — синдромальная форма патологии, включающая сочетание не менее трех

ЭТИОЛОГИЯ
Синдром Алажиля имеет аутосомно-доминантный тип наследования. Генный дефект связан с частичной
ЭТИОЛОГИЯ Синдром Алажиля имеет аутосомно-доминантный тип наследования. Генный дефект связан с частичной

ПАТОГЕНЕЗ
В основе изменений печени при синдроме Алажиля лежит врожденная гипоплазия внутрипеченочных
ПАТОГЕНЕЗ В основе изменений печени при синдроме Алажиля лежит врожденная гипоплазия внутрипеченочных

КЛИНИЧЕСКАЯ КАРТИНА
Синдром холестаза появляется в период новорожденности, реже в течение первых
КЛИНИЧЕСКАЯ КАРТИНА Синдром холестаза появляется в период новорожденности, реже в течение первых

ДИАГНОСТИКА
Физикальное исследование
Необходимо оценить цвет кожного покрова и склер, размеры печени и
ДИАГНОСТИКА Физикальное исследование Необходимо оценить цвет кожного покрова и склер, размеры печени и

КЛИНИЧЕСКАЯ КАРТИНА
Возможны два варианта течения болезни.
При легком варианте отмечают купирование клинических
КЛИНИЧЕСКАЯ КАРТИНА Возможны два варианта течения болезни. При легком варианте отмечают купирование клинических

Изменения глаз
Наиболее типичное изменение — задний эмбриотоксон (малая аномалия развития в
Изменения глаз Наиболее типичное изменение — задний эмбриотоксон (малая аномалия развития в

ЛЕЧЕНИЕ
Патогенетическое.
Цели лечения: коррекция осложнений длительно сохраняющегося холестаза.
Немедикаментозное лечение
Лечебное питание с повышенным
ЛЕЧЕНИЕ Патогенетическое. Цели лечения: коррекция осложнений длительно сохраняющегося холестаза. Немедикаментозное лечение Лечебное питание с повышенным

Дефицит А1АТ
Аутосомно-рецессивное заболевание, вызываемое нарушением синтеза альфа 1-антитрипсина
Пониженная активность А1АТ в
Дефицит А1АТ
Аутосомно-рецессивное заболевание, вызываемое нарушением синтеза альфа 1-антитрипсина
Пониженная активность А1АТ в

Альфа-1-антитрипсин – белок, который вырабатывается печенью. Он помогает организму в
Альфа-1-антитрипсин – белок, который вырабатывается печенью. Он помогает организму в

Синтез альфа-1-антитрипсина регулируется двумя копиями гена протеазного ингибитора серпина-1. Это так
Синтез альфа-1-антитрипсина регулируется двумя копиями гена протеазного ингибитора серпина-1. Это так

А1АТ
Представитель семейства серпинов
Серпины являются ингибиторами сериновых протеаз
Основная функция – ингибирование эластазы
Эластаза
А1АТ
Представитель семейства серпинов
Серпины являются ингибиторами сериновых протеаз
Основная функция – ингибирование эластазы
Эластаза

А1АТ: ген
SERPINA1 (или Pi)
14q32.1
12,2 kbp
7 экзонов (4 кодирующих, 3 некодирующих), 6
А1АТ: ген
SERPINA1 (или Pi)
14q32.1
12,2 kbp
7 экзонов (4 кодирующих, 3 некодирующих), 6

Количество производимого альфа-1-антитрипсина и его активность зависят от типа унаследованной
Количество производимого альфа-1-антитрипсина и его активность зависят от типа унаследованной
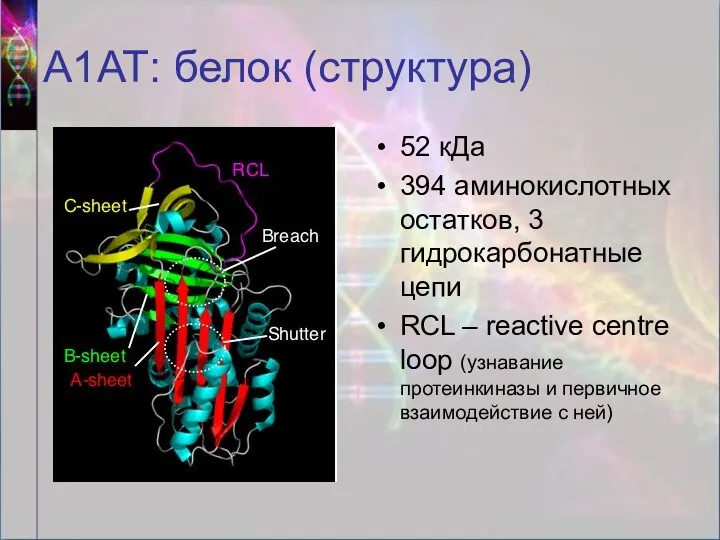
А1АТ: белок (структура)
52 кДа
394 аминокислотных остатков, 3 гидрокарбонатные цепи
RCL – reactive
А1АТ: белок (структура)
52 кДа
394 аминокислотных остатков, 3 гидрокарбонатные цепи
RCL – reactive

А1АТ: белок (механизм ингибирования) (1)
RCL ковалентно связывается с протеазой
Конформационные изменения
А1АТ: белок (механизм ингибирования) (1)
RCL ковалентно связывается с протеазой
Конформационные изменения

А1АТ: белок (механизм ингибирования) (2)
Протеаза атакует RCL
RCL встраивается в бета-лист А,
А1АТ: белок (механизм ингибирования) (2)
Протеаза атакует RCL
RCL встраивается в бета-лист А,

А1АТ: мутации
Приводят к неправильному фолдингу, полимеризации, связыванию двух А1АТ друг с
А1АТ: мутации
Приводят к неправильному фолдингу, полимеризации, связыванию двух А1АТ друг с

Показания к опредению А1АТ:
Если желтуха у новорождённого или малолетнего ребенка длится
Показания к опредению А1АТ:
Если желтуха у новорождённого или малолетнего ребенка длится

Эмфизема
Недостаток А1АТ (нормальное содержание – 1,5-3,5 г/л) => неконтролируемая активность протеаз,
Эмфизема
Недостаток А1АТ (нормальное содержание – 1,5-3,5 г/л) => неконтролируемая активность протеаз,

Диагностирование
95% случаев – не диагностировано
Содержание А1АТ в сыворотке или плазме крови
Диагностирование
95% случаев – не диагностировано
Содержание А1АТ в сыворотке или плазме крови

Лечение
Лечение симптомов
Регулярное введение А1АТ (внутривенные инфузии из донорной плазмы крови,
Лечение
Лечение симптомов
Регулярное введение А1АТ (внутривенные инфузии из донорной плазмы крови,

Болезнь Байлера – это редкое наследственное (передающееся от родителей к детям) заболевание,
Болезнь Байлера – это редкое наследственное (передающееся от родителей к детям) заболевание,

Симптомы болезнь байлера
Заболевание проявляется синдромом холестаза. Для него характерен ряд симптомов.
Главным симптомом холестаза является
Симптомы болезнь байлера
Заболевание проявляется синдромом холестаза. Для него характерен ряд симптомов.
Главным симптомом холестаза является

Причины
В основе синдрома и болезни Байлера лежит нарушение транспорта (переноса) желчных
Причины
В основе синдрома и болезни Байлера лежит нарушение транспорта (переноса) желчных

Диагностика
Анализ анамнеза заболевания и жалоб пациента (когда (как давно) появились желтуха
Диагностика
Анализ анамнеза заболевания и жалоб пациента (когда (как давно) появились желтуха

Значения PGA колеблются от 0 до 12. Если PGA<2, вероятность цирроза
Значения PGA колеблются от 0 до 12. Если PGA<2, вероятность цирроза

Лечение болезнь байлера
Выделяют консервативное (безоперационное) и хирургическое лечение заболевания, а также общие рекомендации.
К общим рекомендациям относится диетотерапия.
Стол №5 по
Лечение болезнь байлера
Выделяют консервативное (безоперационное) и хирургическое лечение заболевания, а также общие рекомендации.
К общим рекомендациям относится диетотерапия.
Стол №5 по

Муковисцидоз
- наследственная болезнь, характеризующаяся системным поражением экзокринных желез (внешней секреции)
Муковисцидоз
- наследственная болезнь, характеризующаяся системным поражением экзокринных желез (внешней секреции)

Из-за нарушения транспорта электролитов через мембрану клеток, которые выстилают протоки желез
Из-за нарушения транспорта электролитов через мембрану клеток, которые выстилают протоки желез

I. Формы муковисцидоза
• Смешанная (легочно-кишечная) с поражением желудочно-кишечного тракта и бронхолегочной
I. Формы муковисцидоза • Смешанная (легочно-кишечная) с поражением желудочно-кишечного тракта и бронхолегочной

симптомы со стороны дыхательной системы: хронический кашель, рецидивирующие пневмонии и ателектазы, перерастяжение
симптомы со стороны дыхательной системы: хронический кашель, рецидивирующие пневмонии и ателектазы, перерастяжение

При диагностике муковисцидоза учитываются данные клиники, истории развития ребенка и семьи,

Недостаток витамина Е проявляется гемолитической анемией у новорожденных и неврологической симптоматикой
Недостаток витамина Е проявляется гемолитической анемией у новорожденных и неврологической симптоматикой

Сахарный диабет выявляется у 20% взрослых пациентов с муковисцидозом.
Фиброз печени, развивающийся
Сахарный диабет выявляется у 20% взрослых пациентов с муковисцидозом.
Фиброз печени, развивающийся
 Патология эмоциональной сферы. Расстройства влечений. Патология двигательной и волевой сферы
Патология эмоциональной сферы. Расстройства влечений. Патология двигательной и волевой сферы Выделительная система
Выделительная система Ауыз қуыс кілегей қабық ауруларына тағайындалатын дәрілік терпияның салыстырмалы сипаттамасы
Ауыз қуыс кілегей қабық ауруларына тағайындалатын дәрілік терпияның салыстырмалы сипаттамасы Дисциркуляторная энцефалопатия и болезнь мелких сосудов
Дисциркуляторная энцефалопатия и болезнь мелких сосудов Жүкті әйелдердегі ауруханадан тыс пневмония
Жүкті әйелдердегі ауруханадан тыс пневмония Анафилаксикалық шок кезіндегі жедел көмек
Анафилаксикалық шок кезіндегі жедел көмек Рак тела матки
Рак тела матки Краснуха - острая вирусная антропонозная инфекция
Краснуха - острая вирусная антропонозная инфекция Нематодозы. Аскаридоз
Нематодозы. Аскаридоз Аномалии количества зубов
Аномалии количества зубов Операции на органах шеи
Операции на органах шеи Методы диагностики гортани
Методы диагностики гортани Легочная гипертензия
Легочная гипертензия Сестринский уход за пациентами с хронической сердечной недостаточностью
Сестринский уход за пациентами с хронической сердечной недостаточностью Лечение открытых переломов
Лечение открытых переломов Збудники шигельозів, холери
Збудники шигельозів, холери Роль гормонов в регуляции роста, развития и гомеостаза
Роль гормонов в регуляции роста, развития и гомеостаза Тыныс алу ағзаларының аурулары
Тыныс алу ағзаларының аурулары Тері физиологиясы
Тері физиологиясы Бустеры Expert Boost. Уход за кожей
Бустеры Expert Boost. Уход за кожей Таным туралы ілім. Медициналық, ғылыми танымның ерекшелігі
Таным туралы ілім. Медициналық, ғылыми танымның ерекшелігі Placental abruption
Placental abruption Альбинизм. Типы альбинизма
Альбинизм. Типы альбинизма Современные аспекты сердечно-легочной и церебральной реанимации в стоматологической практике
Современные аспекты сердечно-легочной и церебральной реанимации в стоматологической практике Факторы риска при инфакте миокарда в развитии низкой физической активности найти информацию научных статей
Факторы риска при инфакте миокарда в развитии низкой физической активности найти информацию научных статей Балалардағы бұлшық ет жүйесінің анатомо-физиологиялық ерекшеліктері
Балалардағы бұлшық ет жүйесінің анатомо-физиологиялық ерекшеліктері Профилактическая медицина
Профилактическая медицина Аккредитация медицинских работников январь 2023
Аккредитация медицинских работников январь 2023