- Генетические заболевания печени в практике врача

Содержание
- 2. Слова французского ученого Жана Доссе, лауреата Нобелевской премии за открытие главного локуса гистосовместимости человека (HLA), о
- 3. Первичная печеночная манифестация Первичная внепеченочная манифестация Генетические заболевания печени Burdelski et al. (1991)
- 4. Генетические заболевания печени с первичной печеночной манифестацией Болезнь Вильсона Наследственный гемохроматоз Дефицит α 1-антитрипсина Кистозный фиброз
- 5. ДНК T A A A G C G A C A T 3' 5' 3' 5'
- 6. Методы диагностики основные методы диагностики хромосомных болезней – цитогенетический анализ), методы диагностики генных болезней (молекулярно–генетический анализ)
- 7. Болезни накопления Болезнь Вильсона Наследственный гемохроматоз Дефицит α 1-антитрипсина Гликогенозы При уточнении генеза заболеваний печени у
- 8. Пациент С. 26 лет Жалобы: на незначительную слабость, повышение t до 37,1-37,2 С̥ в вечерние часы.
- 9. Пациент С. 26 лет Анамнез: болен в течение года, когда обратил внимание на слабость, недомогание, повышение
- 10. Пациент С .26 лет Предварительный диагноз? Рекомендуемое обследование?
- 11. Болезнь Вильсона- Коновалова Аутосомно-рециссивный тип наследования Токсическое накопление меди Частота: 1:30.000; частота аллелей 1:90 Мутация гена
- 12. Обмен Cu++ в организме Cu++ транспортируется в эпителий проксимальной части тонкой кишки, часть (40-75%) остается в
- 13. Патогенез болезни Вильсона Снижение экскреции Сu++ с желчью из гепатоцита, обусловленное дефицитом или полным отсутствием продукта
- 14. Токсическое влияние меди в печени Продукция свободных радикалов Токсическое влияние на лизосомы Токсическое влияние на митохондрии
- 15. Механизмы токсического действия Cu++ Cu++- прооксидант катализирует образование свободных радикалов и запускает процесс ПОЛ: - нарушается
- 16. Проявления болезни Вильсона Maнифестация Симптомы Печень. Стеатоз печени, фиброз, цирроз с портальной гипертензией, хронический активный гепатит,фульминантная
- 17. Клинические проявления болезни Вильсона Дети 4-5 лет печеночная манифестация (у 42% больных). 20-30 лет - Нейропсихическая
- 18. Формы поражения печени при болезни Вильсона. Острый гепатит - 25% ( маска – инфекционный гепатит: желтуха,
- 19. Формы поражения печени при болезни Вильсона (продолжение) Цирроз печени – протекает бессимптомно или малосимптомно. Выявляется у
- 20. Подозрение на Болезнь Вильсона. Возраст менее 40 лет при наличии: - необъяснимых расстройств ЦНС + признаков
- 21. Скрининг на наличие Болезни Вильсона концентрация церулоплазмина в сыворотке крови кольцо Кайзера-Флейшера
- 22. Диагностика б-ни Вильсона Осмотр в щелевой лампе ( кольцо Кайзера Флейшера) Церулоплазмин в сыворотке крови Медь
- 23. Подтверждение Болезни Вильсона Суточная экскреция Сu++ с мочой >100мкг/сутки Биопсия печени с определением Сu++ в ткани
- 24. Кольцо Кайзера Флейшера
- 25. Кольцо Кайзера Флешнера
- 26. Катаракта «подсолнечник»
- 27. Болезнь Вильсона : гистология печени Гематоксилин-эозин(краска) Роданин (окраска)
- 28. Генетическое исследование при б-ни Вильсона норма c.778_779insC (Insertion of the Cytosine Base Following Nucleotide 778 in
- 29. Лечение Болезни Вильсона Диета (исключение из рациона продуктов с высоким содержанием Cu: необработанная пшеница, бобы, горох,
- 30. Б-нь Вильсона: Медикаментозная терапия Лекарство Дозы Д-Пеницилламин 1.0 – 1.5 г/день (взрослые) 20 мг/кг/день (дети) Триентин
- 31. Д-пеницилламин (купренил) – золотой стандарт лечения б-ни Вильсона 1 этап- начальная доза 250-500 мг 1-2 раза
- 32. Побочные эффекты Д-пеницилламина (ранние : 1-ый месяц лечения) Ухудшение неврологической симптоматики (Сu уходит из депо печени
- 33. Побочные эффекты Д-пеницилламина (поздние) Кожные изменения : пеницилламиновая дерматопатия, пемфигус (группа различных по своей природе заболеваний,
- 34. acantosis nigricans . (чёрный акантоз, дистрофия кожи пигментно-сосочковая)
- 35. Другие лекарственные препараты для лечения Болезни Вильсона Триентин- альтернативный медьхелатирующий препарат. Используется с 1969 года у
- 37. Пациентка К. 26 лет Дата. 12.08.2008г. Жалобы на ощущение жжения и зуд кистей. Анамнез: летом 2006
- 38. Данные объективного осмотра: Кожные покровы бледно-розовые, умеренно-влажные, чистые. Подкожно-жировая клетчатка развита умеренно. Лимфатические узлы не увеличены,
- 39. Лабораторные исследования: Общий анализ крови: Hb.- 129г/л ,Эритр.- 5.0х1012, Тромб.- 125х109,Лейк.- 6,5х 109; п/я- 2; с/я-
- 40. Инструментальные исследования: УЗИ: Печень не увеличена: левая доля 58х73 мм, правая доля- 121х132 мм,хвостатая доля –
- 41. Дополнительные исследования: Церулоплазмин: 15 мг/дл (N = 20-60). Медь в плазме крови -0,33 мг/л (N =
- 42. Клинический диагноз: Болезнь Вильсона –Коновалова, висцеральная форма : цирроз печени с синдромом портальной гипертензии (спленомегалия ВРВП
- 43. Рекомендации: Полное исключение алкоголя, инсоляций, вакцинаций, необоснованного приема лекарств Из рациона исключить продукты, богатые медью.: орехи,
- 44. Кольцо Кайзера-Флейшера до (снимок справа) и после (снимок слева) лечения
- 45. Б-нь Вильсона Выживаемость после трансплантации печени 87 86 84 82 79 0 10 20 30 40
- 46. Гемохроматоз
- 47. Основные показатели обмена Fe в организме Всего 4-5 г: Гемоглобин ~ 50% Скелетные мышцы – 15%
- 48. Обмен железа 1-2 мг железа в сутки всасывается в 12ПК и попадают в плазму в зависимости
- 49. Синдром перегрузки железом Первичное увеличение содержания железа в крови, связанное с наследственным дефектом метаболизма, вследствие которого
- 50. Состояния, сопровождающиеся повышением накопления железа клетками печени Наследственный гемохроматоз. - HFE ассоциированный [Мутация С282Y/C282Y. Мутация С282Y/H63D.
- 51. Состояния, сопровождающиеся повышением накопления железа клетками печени Вторичный гемохроматоз: -Приобретенное увеличение накопления железа клетками печени (анемии,
- 52. ИДИОПАТИЧЕСКИЙ ГЕМОХРОМАТОЗ наследственно обусловленное первичное расстройство обмена Fe, сопровождающееся повышенной абсорбцией Fe в кишечнике и первичным
- 53. Hаследственный гемохроматоз («Бронзовый диабет», «Пигментный цирроз») Аутосомно-рецессивный тип наследования Диагноз обычно подтверждается через 5–10 лет после
- 54. Состояния, сопровождающиеся повышением накопления железа клетками печени Наследственный гемохроматоз. - HFE ассоциированный [Мутация С282Y/C282Y (87-90%). Мутация
- 55. Наследственный гемохроматоз: HFE протеин- триггер генетического заболевания. Физиологическая регуляция Fe DMT1 Ферритин Ферропортин Трансферрин HFE HFE
- 56. Наследственный гемохроматоз: HFE протеин- триггер генетического заболевания. Патофизиология регуляции Fe при дефиците HFE Энтероцит DMT1 Ферритин
- 57. Механизмы тканевого повреждения Разрушение Fe-«загруженных» лизосом Пероксидация липидов внутриклеточных органелл Стимуляция синтеза коллагена
- 58. Механизмы тканевого повреждения Разрушение Fe-«загруженных» лизосом Пероксидация липидов внутриклеточных органелл Стимуляция синтеза коллагена (активация звездчатых клеток)
- 59. Предикторы значимого фиброза Мужской пол (для групп Длительность вирусной инфекции (преимущественно гепатита С) Ожирение, сахарный диабет
- 60. Наследственный гемохроматоз (Накопление Fe) Концентрация ферритина в крови 0 500 1000 1500 Возраст [лет] 0 10
- 61. Клинические проявления гемохроматоза Начальные симптомы гемохроматоза: Слабость Утомляемость Потеря веса Изменения окраски кожи (дымчатая) Боли в
- 62. Методы, характеризующие избыточное накопление железа Концентрация железа и ферритина в сыворотке крови Процент насыщения трансферрина -
- 63. Наследственный гемохроматоз: гистологическая картина : Prussian blue
- 64. Клинические тесты ИГХ в порядке уменьшения их чувствительности НТЖ ( >45% или >35% в предклимактерическом периоде
- 65. Наследственный гемохроматоз: Диагностический алгоритм. Наследств. гемохроматоз подтвержден Наследств. гемохроматоз подтвержден Биопсия печени Мониторирование Неизвестное заболевание печени
- 66. Наследственный гемохроматоз: рекомендации по диете Запрет на введение железа Умеренное потребление мяса Избегать употребления алкоголя Ограничение
- 67. ЛЕЧЕНИЕ ГЕМОХРОМАТОЗА Диета Исключить продукты с высоким содержанием Fe: сушеные белые грибы, печень и почки, персики,
- 68. Лечебные мероприятия, способствующие регрессу фиброза печени Прекращение приема алкоголя при алкогольных поражениях печени Длительная иммуносупрессивная терапия
- 69. Наследственный гемохроматоз: терапия Начально, еженедельно удаление при флеботомии до 500 мл крови (500 ml соответствуют 250
- 70. Лечение гемохроматоза Дефероксамин - комплексобразователь. Менее эффективен, чем венесекция. Используется при тяжелой кардиомиопатии и нарушении ритма
- 71. Лечение гемохроматоза Деферазирокс (Эксиджад) – таблетки по 250 и 500 мг. Представляет собой тридентатный лиганд, который
- 72. Причины смерти больных ИГХ Сердечная недостаточность ~ 30% Печеночная недостаточность или портальная гипертензия ~ 25% Гепатоцеллюлярная
- 73. Data from European Liver Transplant Association (www.eltr.org) 6/2006 Наследственный гемохроматоз: Выживаемость после трансплантации печени 74 71
- 74. Наследственный гемохроматоз: заключение Наследственный гемахроматоз – одно из наиболее распространенных генетических заболеваний печени Молекулярно-генетические исследований необходимо
- 76. Скачать презентацию

Слова французского ученого Жана Доссе, лауреата Нобелевской премии за открытие главного
Слова французского ученого Жана Доссе, лауреата Нобелевской премии за открытие главного
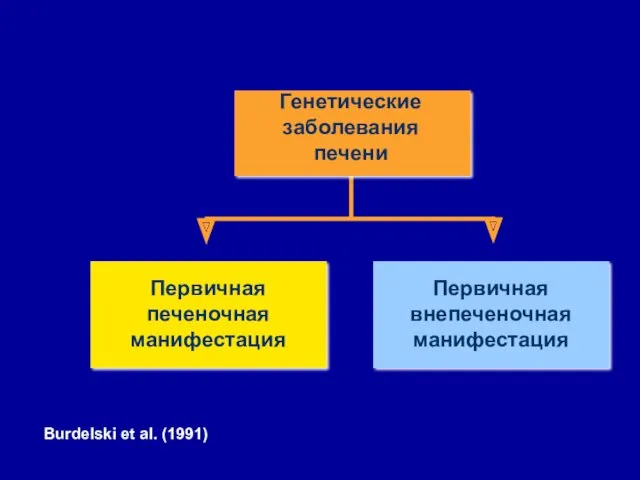
Первичная печеночная манифестация
Первичная внепеченочная манифестация
Генетические заболевания печени
Burdelski et al. (1991)
Первичная печеночная манифестация
Первичная внепеченочная манифестация
Генетические заболевания печени
Burdelski et al. (1991)

Генетические заболевания печени с первичной печеночной манифестацией
Болезнь Вильсона
Наследственный гемохроматоз
Дефицит α 1-антитрипсина
Кистозный
Генетические заболевания печени с первичной печеночной манифестацией
Болезнь Вильсона
Наследственный гемохроматоз
Дефицит α 1-антитрипсина
Кистозный
ДНК
T
A
A
A
G
C
G
A
C
A
T
3'
5'
3'
5'
T
T
A
C
C
T
T
T
G
G
A
5'
3'
U
A
A
A
G
C
G
A
C
A
U
mРНК
Транскрипция
Tрансляция
Белок
NH2
COOH
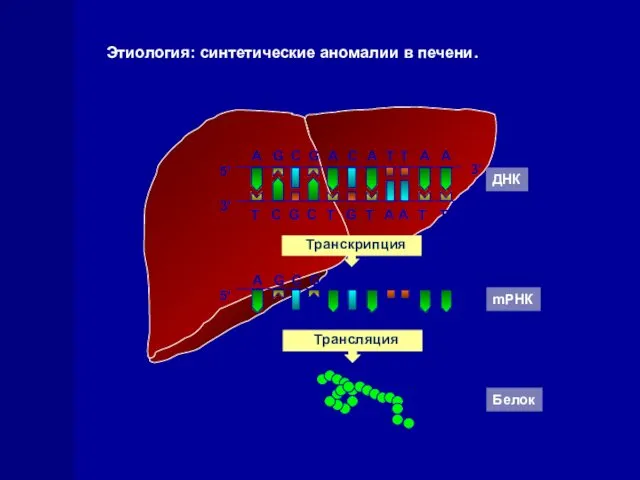
Этиология: синтетические аномалии в печени.
ДНК
T
A
A
A
G
C
G
A
C
A
T
3'
5'
3'
5'
T
T
A
C
C
T
T
T
G
G
A
5'
3'
U
A
A
A
G
C
G
A
C
A
U
mРНК
Транскрипция
Tрансляция
Белок
NH2
COOH
Этиология: синтетические аномалии в печени.

Методы диагностики
основные методы диагностики хромосомных болезней – цитогенетический анализ),
методы диагностики
Методы диагностики
основные методы диагностики хромосомных болезней – цитогенетический анализ),
методы диагностики

Болезни накопления
Болезнь Вильсона
Наследственный гемохроматоз
Дефицит α 1-антитрипсина
Гликогенозы
При уточнении генеза заболеваний печени у
Болезни накопления
Болезнь Вильсона
Наследственный гемохроматоз
Дефицит α 1-антитрипсина
Гликогенозы
При уточнении генеза заболеваний печени у

Пациент С. 26 лет
Жалобы: на незначительную слабость, повышение t до 37,1-37,2
Пациент С. 26 лет
Жалобы: на незначительную слабость, повышение t до 37,1-37,2

Пациент С. 26 лет
Анамнез: болен в течение года, когда обратил внимание
Пациент С. 26 лет
Анамнез: болен в течение года, когда обратил внимание

Пациент С .26 лет
Предварительный диагноз?
Рекомендуемое обследование?
Пациент С .26 лет
Предварительный диагноз?
Рекомендуемое обследование?

Болезнь Вильсона- Коновалова
Аутосомно-рециссивный тип наследования
Токсическое накопление меди
Частота: 1:30.000; частота аллелей
Болезнь Вильсона- Коновалова
Аутосомно-рециссивный тип наследования
Токсическое накопление меди
Частота: 1:30.000; частота аллелей

Обмен Cu++ в организме
Cu++ транспортируется в эпителий проксимальной части тонкой
Обмен Cu++ в организме
Cu++ транспортируется в эпителий проксимальной части тонкой

Патогенез болезни Вильсона
Снижение экскреции Сu++ с желчью из гепатоцита, обусловленное дефицитом
Патогенез болезни Вильсона
Снижение экскреции Сu++ с желчью из гепатоцита, обусловленное дефицитом

Токсическое влияние меди в печени
Продукция свободных радикалов
Токсическое влияние на лизосомы
Токсическое влияние
Токсическое влияние меди в печени
Продукция свободных радикалов
Токсическое влияние на лизосомы
Токсическое влияние

Механизмы токсического действия Cu++
Cu++- прооксидант катализирует образование свободных радикалов и запускает
Механизмы токсического действия Cu++
Cu++- прооксидант катализирует образование свободных радикалов и запускает

Проявления болезни Вильсона
Maнифестация Симптомы
Печень. Стеатоз печени, фиброз, цирроз с портальной гипертензией,
хронический
Проявления болезни Вильсона
Maнифестация Симптомы
Печень. Стеатоз печени, фиброз, цирроз с портальной гипертензией,
хронический

Клинические проявления болезни Вильсона
Дети 4-5 лет печеночная манифестация (у 42% больных).
20-30
Клинические проявления болезни Вильсона
Дети 4-5 лет печеночная манифестация (у 42% больных).
20-30

Формы поражения печени при болезни Вильсона.
Острый гепатит - 25% ( маска
Формы поражения печени при болезни Вильсона.
Острый гепатит - 25% ( маска

Формы поражения печени при болезни Вильсона (продолжение)
Цирроз печени – протекает
Формы поражения печени при болезни Вильсона (продолжение)
Цирроз печени – протекает

Подозрение на Болезнь Вильсона.
Возраст менее 40 лет при наличии: - необъяснимых
Подозрение на Болезнь Вильсона.
Возраст менее 40 лет при наличии: - необъяснимых

Скрининг на наличие Болезни Вильсона
концентрация церулоплазмина в сыворотке крови < 20
Скрининг на наличие Болезни Вильсона
концентрация церулоплазмина в сыворотке крови < 20

Диагностика б-ни Вильсона
Осмотр в щелевой лампе ( кольцо Кайзера Флейшера)
Церулоплазмин в
Диагностика б-ни Вильсона
Осмотр в щелевой лампе ( кольцо Кайзера Флейшера)
Церулоплазмин в

Подтверждение Болезни Вильсона
Суточная экскреция Сu++ с мочой >100мкг/сутки
Биопсия печени с
Подтверждение Болезни Вильсона
Суточная экскреция Сu++ с мочой >100мкг/сутки
Биопсия печени с
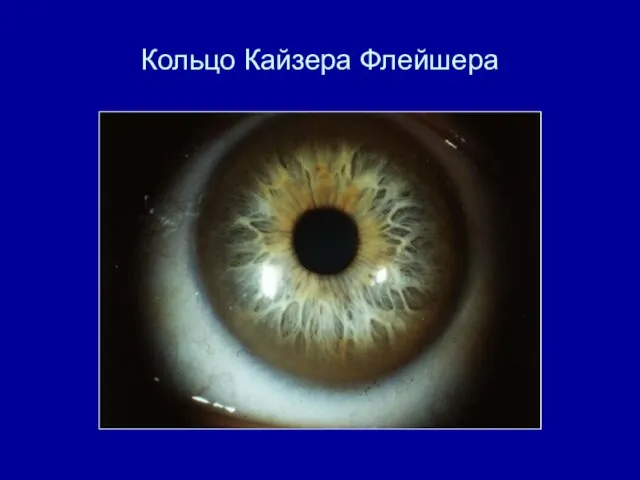
Кольцо Кайзера Флейшера
Кольцо Кайзера Флейшера
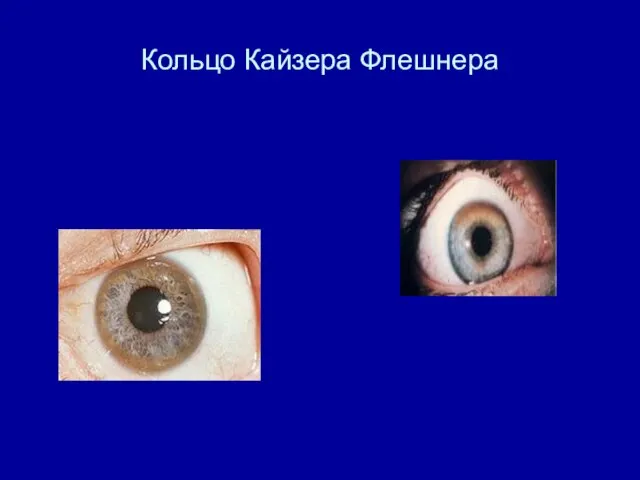
Кольцо Кайзера Флешнера
Кольцо Кайзера Флешнера
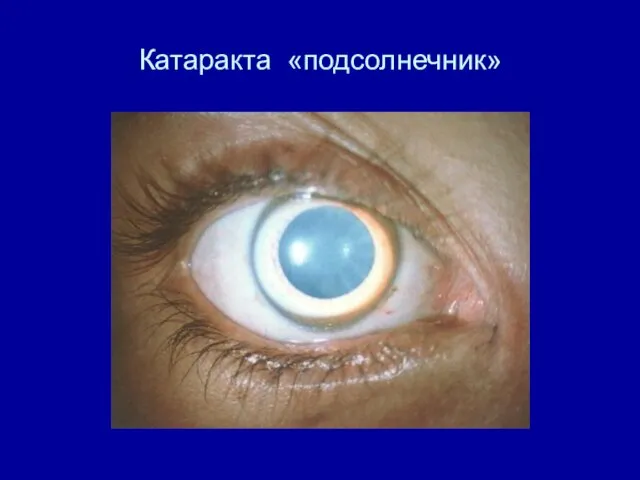
Катаракта «подсолнечник»
Катаракта «подсолнечник»
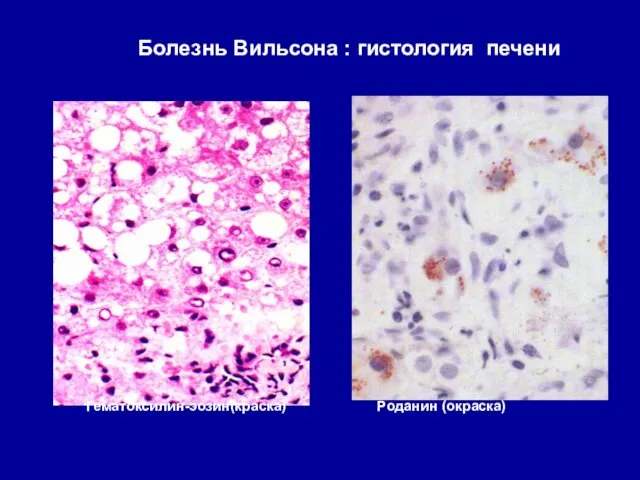
Болезнь Вильсона : гистология печени
Гематоксилин-эозин(краска) Роданин (окраска)
Гематоксилин-эозин(краска) Роданин (окраска)
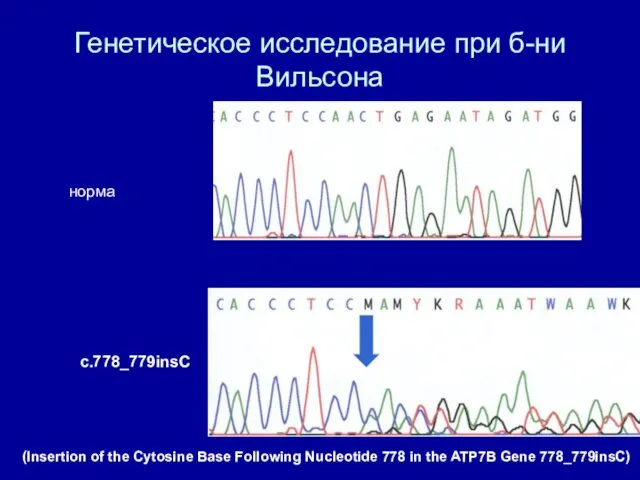
Генетическое исследование при б-ни Вильсона
норма
c.778_779insC
(Insertion of the Cytosine Base Following Nucleotide
Генетическое исследование при б-ни Вильсона
норма
c.778_779insC
(Insertion of the Cytosine Base Following Nucleotide

Лечение Болезни Вильсона
Диета (исключение из рациона продуктов с высоким содержанием
Лечение Болезни Вильсона
Диета (исключение из рациона продуктов с высоким содержанием

Б-нь Вильсона: Медикаментозная терапия
Лекарство
Дозы
Д-Пеницилламин
1.0 – 1.5 г/день (взрослые)
20 мг/кг/день (дети)
Триентин
1.0 –
Лекарство
Дозы
Д-Пеницилламин
1.0 – 1.5 г/день (взрослые)
20 мг/кг/день (дети)
Триентин
1.0 –

Д-пеницилламин (купренил) – золотой стандарт лечения б-ни Вильсона
1 этап- начальная
Д-пеницилламин (купренил) – золотой стандарт лечения б-ни Вильсона
1 этап- начальная

Побочные эффекты Д-пеницилламина (ранние : 1-ый месяц лечения)
Ухудшение неврологической симптоматики (Сu
Побочные эффекты Д-пеницилламина (ранние : 1-ый месяц лечения)
Ухудшение неврологической симптоматики (Сu

Побочные эффекты Д-пеницилламина (поздние)
Кожные изменения : пеницилламиновая дерматопатия, пемфигус (группа различных
Побочные эффекты Д-пеницилламина (поздние)
Кожные изменения : пеницилламиновая дерматопатия, пемфигус (группа различных
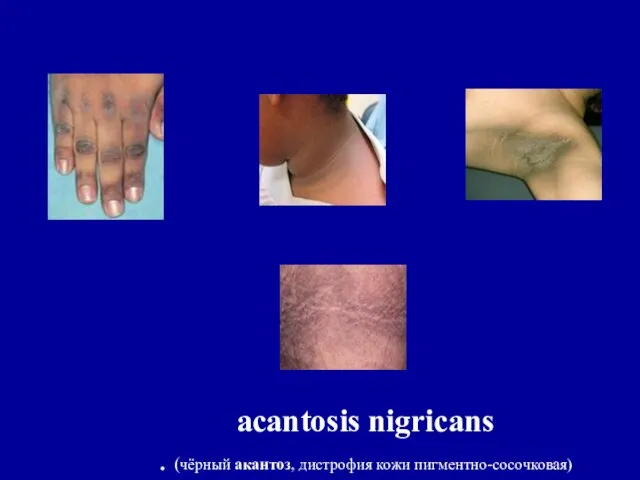
acantosis nigricans
. (чёрный акантоз, дистрофия кожи пигментно-сосочковая)
acantosis nigricans
. (чёрный акантоз, дистрофия кожи пигментно-сосочковая)

Другие лекарственные препараты для лечения Болезни Вильсона
Триентин- альтернативный медьхелатирующий препарат. Используется
Другие лекарственные препараты для лечения Болезни Вильсона
Триентин- альтернативный медьхелатирующий препарат. Используется


Пациентка К. 26 лет
Дата. 12.08.2008г.
Жалобы на ощущение жжения и зуд
Пациентка К. 26 лет
Дата. 12.08.2008г.
Жалобы на ощущение жжения и зуд

Данные объективного осмотра:
Кожные покровы бледно-розовые, умеренно-влажные, чистые. Подкожно-жировая клетчатка развита умеренно.
Данные объективного осмотра:
Кожные покровы бледно-розовые, умеренно-влажные, чистые. Подкожно-жировая клетчатка развита умеренно.

Лабораторные исследования:
Общий анализ крови: Hb.- 129г/л ,Эритр.- 5.0х1012, Тромб.- 125х109,Лейк.- 6,5х
Лабораторные исследования:
Общий анализ крови: Hb.- 129г/л ,Эритр.- 5.0х1012, Тромб.- 125х109,Лейк.- 6,5х

Инструментальные исследования:
УЗИ: Печень не увеличена: левая доля 58х73 мм, правая доля-
Инструментальные исследования:
УЗИ: Печень не увеличена: левая доля 58х73 мм, правая доля-

Дополнительные исследования:
Церулоплазмин: 15 мг/дл (N = 20-60).
Медь в плазме крови -0,33
Дополнительные исследования:
Церулоплазмин: 15 мг/дл (N = 20-60).
Медь в плазме крови -0,33

Клинический диагноз:
Болезнь Вильсона –Коновалова, висцеральная форма : цирроз печени с синдромом
Клинический диагноз:
Болезнь Вильсона –Коновалова, висцеральная форма : цирроз печени с синдромом

Рекомендации:
Полное исключение алкоголя, инсоляций, вакцинаций, необоснованного приема лекарств
Из рациона исключить продукты,
Рекомендации:
Полное исключение алкоголя, инсоляций, вакцинаций, необоснованного приема лекарств
Из рациона исключить продукты,
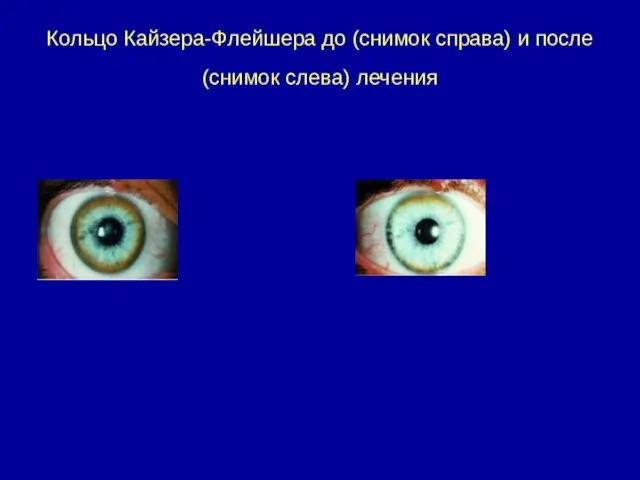
Кольцо Кайзера-Флейшера до (снимок справа) и после (снимок слева) лечения
Кольцо Кайзера-Флейшера до (снимок справа) и после (снимок слева) лечения
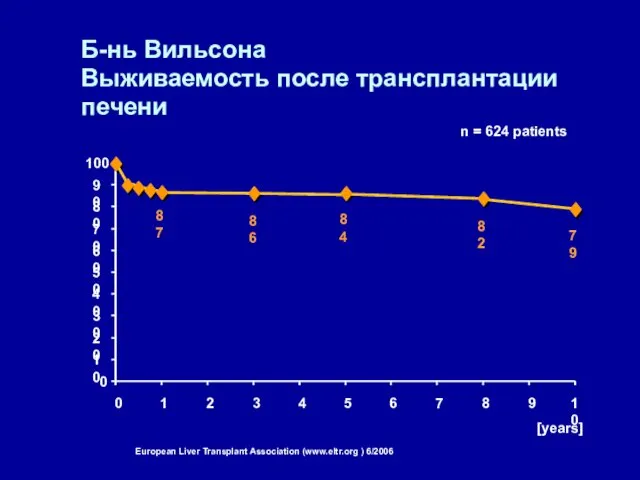
Б-нь Вильсона
Выживаемость после трансплантации печени
87
86
84
82
79
0
10
20
30
40
50
60
70
80
90
100
0
1
2
3
4
5
6
7
8
9
10
[years]
[%] n = 624 patients
Data from
Выживаемость после трансплантации печени
87
86
84
82
79
0
10
20
30
40
50
60
70
80
90
100
0
1
2
3
4
5
6
7
8
9
10
[years]
[%] n = 624 patients
Data from

Гемохроматоз
Гемохроматоз

Основные показатели обмена Fe в организме
Всего 4-5 г:
Гемоглобин ~ 50%
Скелетные мышцы
Основные показатели обмена Fe в организме
Всего 4-5 г:
Гемоглобин ~ 50%
Скелетные мышцы

Обмен железа
1-2 мг железа в сутки всасывается в 12ПК и попадают
Обмен железа
1-2 мг железа в сутки всасывается в 12ПК и попадают

Синдром перегрузки железом
Первичное увеличение содержания железа в крови, связанное с наследственным
Синдром перегрузки железом
Первичное увеличение содержания железа в крови, связанное с наследственным

Состояния, сопровождающиеся повышением накопления железа клетками печени
Наследственный гемохроматоз. - HFE ассоциированный
Состояния, сопровождающиеся повышением накопления железа клетками печени
Наследственный гемохроматоз. - HFE ассоциированный

Состояния, сопровождающиеся повышением накопления железа клетками печени
Вторичный гемохроматоз: -Приобретенное увеличение накопления
Состояния, сопровождающиеся повышением накопления железа клетками печени
Вторичный гемохроматоз: -Приобретенное увеличение накопления

ИДИОПАТИЧЕСКИЙ ГЕМОХРОМАТОЗ
наследственно обусловленное первичное расстройство обмена Fe, сопровождающееся повышенной абсорбцией
ИДИОПАТИЧЕСКИЙ ГЕМОХРОМАТОЗ
наследственно обусловленное первичное расстройство обмена Fe, сопровождающееся повышенной абсорбцией
Hаследственный гемохроматоз («Бронзовый диабет», «Пигментный цирроз»)
Аутосомно-рецессивный тип наследования
Диагноз обычно подтверждается
через 5–10
Аутосомно-рецессивный тип наследования
Диагноз обычно подтверждается
через 5–10

Состояния, сопровождающиеся повышением накопления железа клетками печени
Наследственный гемохроматоз. - HFE ассоциированный
Состояния, сопровождающиеся повышением накопления железа клетками печени
Наследственный гемохроматоз. - HFE ассоциированный
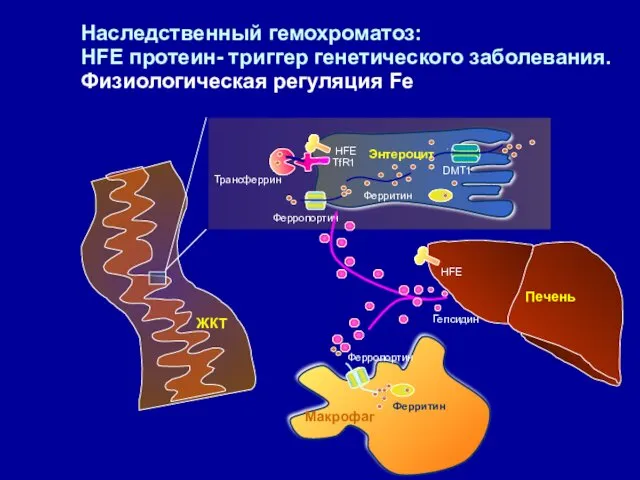
Наследственный гемохроматоз:
HFE протеин- триггер генетического заболевания. Физиологическая регуляция Fe
DMT1
Ферритин
Ферропортин
Трансферрин
HFE
HFE
Печень
Ферритин
Ферропортин
TfR1
Гепсидин
Maкрофаг
ЖКТ
Энтероцит
Наследственный гемохроматоз:
HFE протеин- триггер генетического заболевания. Физиологическая регуляция Fe
DMT1
Ферритин
Ферропортин
Трансферрин
HFE
HFE
Печень
Ферритин
Ферропортин
TfR1
Гепсидин
Maкрофаг
ЖКТ
Энтероцит
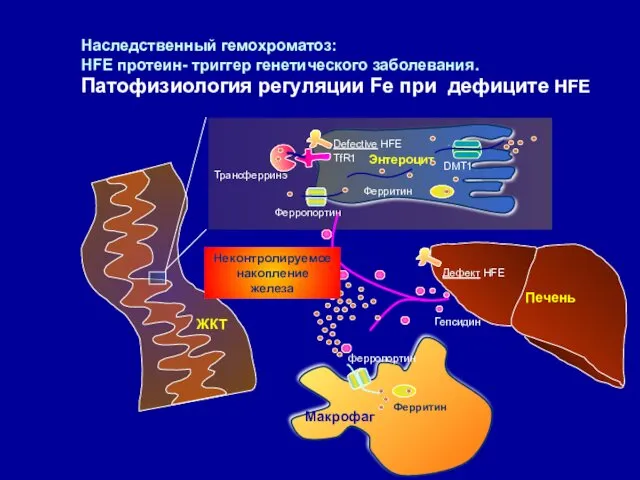
Наследственный гемохроматоз:
HFE протеин- триггер генетического заболевания.
Патофизиология регуляции Fe при дефиците
HFE протеин- триггер генетического заболевания.
Патофизиология регуляции Fe при дефиците

Механизмы тканевого повреждения
Разрушение Fe-«загруженных» лизосом
Пероксидация липидов внутриклеточных органелл
Стимуляция синтеза коллагена
Механизмы тканевого повреждения
Разрушение Fe-«загруженных» лизосом
Пероксидация липидов внутриклеточных органелл
Стимуляция синтеза коллагена
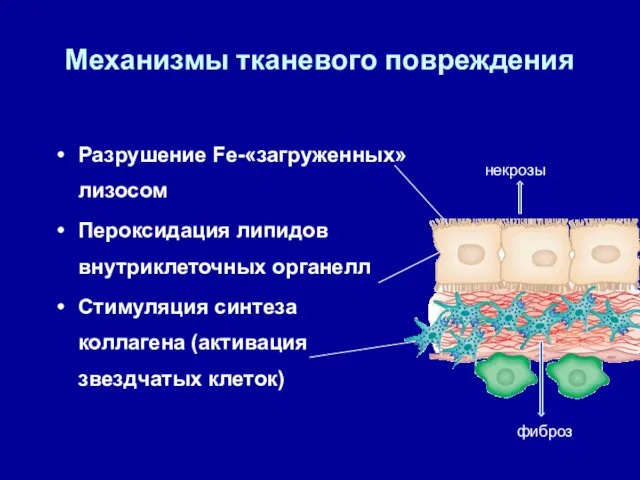
Механизмы тканевого повреждения
Разрушение Fe-«загруженных» лизосом
Пероксидация липидов внутриклеточных органелл
Стимуляция синтеза коллагена (активация
Механизмы тканевого повреждения
Разрушение Fe-«загруженных» лизосом
Пероксидация липидов внутриклеточных органелл
Стимуляция синтеза коллагена (активация

Предикторы значимого фиброза
Мужской пол (для групп < 50 лет)
Длительность вирусной инфекции
Предикторы значимого фиброза
Мужской пол (для групп < 50 лет)
Длительность вирусной инфекции
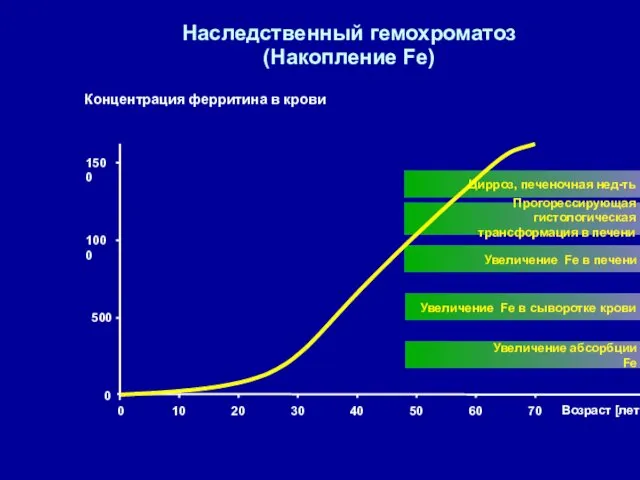
Наследственный гемохроматоз
(Накопление Fe)
Концентрация ферритина в крови
0
500
1000
1500
Возраст [лет]
0
10
20
30
40
50
60
70
Увеличение абсорбции Fe
Увеличение Fe
Наследственный гемохроматоз
(Накопление Fe)
Концентрация ферритина в крови
0
500
1000
1500
Возраст [лет]
0
10
20
30
40
50
60
70
Увеличение абсорбции Fe
Увеличение Fe

Клинические проявления гемохроматоза
Начальные симптомы гемохроматоза: Слабость Утомляемость Потеря веса Изменения окраски
Клинические проявления гемохроматоза
Начальные симптомы гемохроматоза: Слабость Утомляемость Потеря веса Изменения окраски

Методы, характеризующие избыточное накопление железа
Концентрация железа и ферритина в сыворотке крови
Процент
Методы, характеризующие избыточное накопление железа
Концентрация железа и ферритина в сыворотке крови
Процент
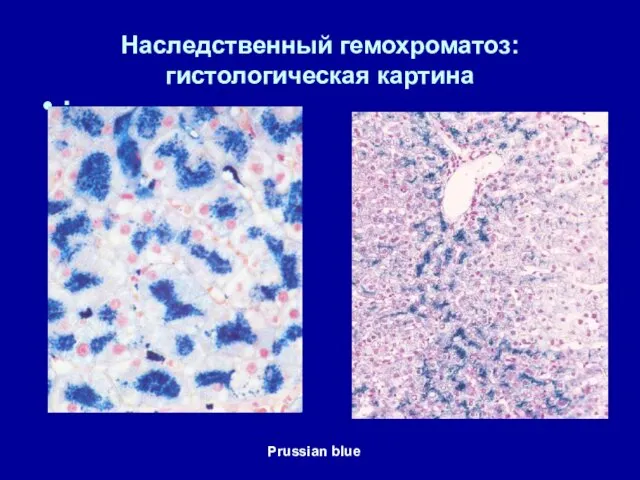
Наследственный гемохроматоз:
гистологическая картина
:
Prussian blue
Наследственный гемохроматоз:
гистологическая картина
:
Prussian blue

Клинические тесты ИГХ в порядке уменьшения их чувствительности
НТЖ ( >45% или
Клинические тесты ИГХ в порядке уменьшения их чувствительности
НТЖ ( >45% или
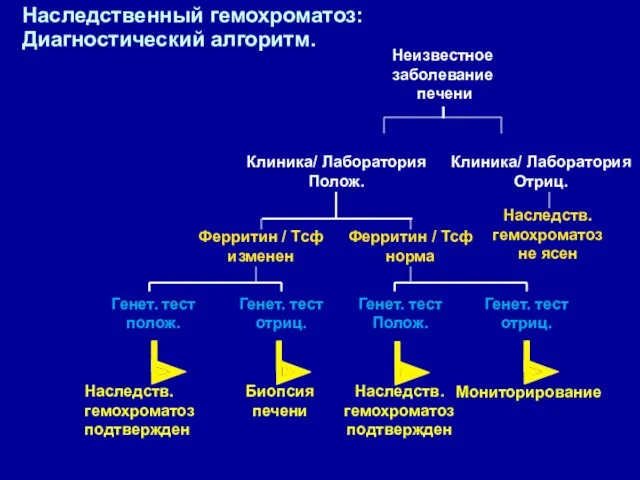
Наследственный гемохроматоз:
Диагностический алгоритм.
Наследств.
гемохроматоз
подтвержден
Наследств.
гемохроматоз
подтвержден
Биопсия
печени
Мониторирование
Неизвестное
заболевание
печени
l
Клиника/ Лаборатория
Полож.
Клиника/ Лаборатория
Отриц.
Наследств.
Наследственный гемохроматоз:
Диагностический алгоритм.
Наследств.
гемохроматоз
подтвержден
Наследств.
гемохроматоз
подтвержден
Биопсия
печени
Мониторирование
Неизвестное
заболевание
печени
l
Клиника/ Лаборатория
Полож.
Клиника/ Лаборатория
Отриц.
Наследств.

Наследственный гемохроматоз: рекомендации по диете
Запрет на введение железа
Умеренное потребление мяса
Избегать употребления
Наследственный гемохроматоз: рекомендации по диете
Запрет на введение железа
Умеренное потребление мяса
Избегать употребления

ЛЕЧЕНИЕ ГЕМОХРОМАТОЗА
Диета
Исключить продукты с высоким содержанием Fe: сушеные белые грибы, печень
ЛЕЧЕНИЕ ГЕМОХРОМАТОЗА
Диета Исключить продукты с высоким содержанием Fe: сушеные белые грибы, печень

Лечебные мероприятия, способствующие регрессу фиброза печени
Прекращение приема алкоголя при алкогольных поражениях
Лечебные мероприятия, способствующие регрессу фиброза печени
Прекращение приема алкоголя при алкогольных поражениях

Наследственный гемохроматоз: терапия
Начально, еженедельно удаление при флеботомии до 500 мл крови
(500
Наследственный гемохроматоз: терапия
Начально, еженедельно удаление при флеботомии до 500 мл крови
(500

Лечение гемохроматоза
Дефероксамин - комплексобразователь. Менее эффективен, чем венесекция. Используется при тяжелой
Лечение гемохроматоза
Дефероксамин - комплексобразователь. Менее эффективен, чем венесекция. Используется при тяжелой

Лечение гемохроматоза
Деферазирокс (Эксиджад) – таблетки по 250 и 500 мг. Представляет
Лечение гемохроматоза
Деферазирокс (Эксиджад) – таблетки по 250 и 500 мг. Представляет

Причины смерти больных ИГХ
Сердечная недостаточность ~ 30%
Печеночная недостаточность или портальная гипертензия
Причины смерти больных ИГХ
Сердечная недостаточность ~ 30%
Печеночная недостаточность или портальная гипертензия
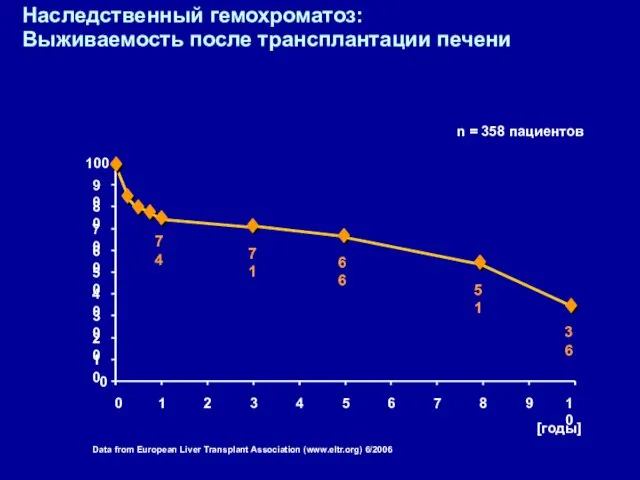
Data from European Liver Transplant Association (www.eltr.org) 6/2006
Наследственный гемохроматоз:
Выживаемость после трансплантации
Data from European Liver Transplant Association (www.eltr.org) 6/2006
Наследственный гемохроматоз:
Выживаемость после трансплантации

Наследственный гемохроматоз: заключение
Наследственный гемахроматоз – одно из наиболее распространенных генетических заболеваний
Наследственный гемохроматоз: заключение
Наследственный гемахроматоз – одно из наиболее распространенных генетических заболеваний
 Анимированный кроссворд Первоначальные химические понятия
Анимированный кроссворд Первоначальные химические понятия Фигура человека в движении. Этапы рисования
Фигура человека в движении. Этапы рисования Теория ландшафтной архитектуры и методология проектирования
Теория ландшафтной архитектуры и методология проектирования Философия бытия. Модуль 3
Философия бытия. Модуль 3 Проект Математика вокруг нас
Проект Математика вокруг нас Религиозный состав России. Интерактивный тест
Религиозный состав России. Интерактивный тест Чудо Болдинской осени. Теперь моя пора. А.С. Пушкин
Чудо Болдинской осени. Теперь моя пора. А.С. Пушкин Видільна система. 8 клас
Видільна система. 8 клас Педагогическая мастерская:Развитие креативного мышления творчески одарённых детей (презентация)
Педагогическая мастерская:Развитие креативного мышления творчески одарённых детей (презентация) Проектирование образовательной среды МБОУ СОШ №1,
Проектирование образовательной среды МБОУ СОШ №1, Страхование ответственности за загрязнение окружающей среды (экологическое страхование) в России и за рубежом
Страхование ответственности за загрязнение окружающей среды (экологическое страхование) в России и за рубежом презентация Турнир по географии 7 класс
презентация Турнир по географии 7 класс Нелинейные электрические цепи постоянного тока
Нелинейные электрические цепи постоянного тока День учителя
День учителя Дидактические игры по УМК своими руками
Дидактические игры по УМК своими руками Фермы металлодеревянные треугольные
Фермы металлодеревянные треугольные Отчет летнего студенческого отряда ГГХПИ за июль-август 2012 года
Отчет летнего студенческого отряда ГГХПИ за июль-август 2012 года дистант по географии 8 класс
дистант по географии 8 класс Электронно-дырочный переход. Определение и классификация
Электронно-дырочный переход. Определение и классификация Сочинение-рассуждение на лингвистическую тему
Сочинение-рассуждение на лингвистическую тему Управление состоянием откосов. Лекция 15
Управление состоянием откосов. Лекция 15 Формулы сокращенного умножения
Формулы сокращенного умножения Виды, разрезы, сечения
Виды, разрезы, сечения Атлантический океан
Атлантический океан Богатства Южного Урала
Богатства Южного Урала Курсовой проект Кадет. Курсант. Офицер
Курсовой проект Кадет. Курсант. Офицер Directions, part 3
Directions, part 3 презентация к родительскому собранию Агрессия детей, ее причины и предупреждение
презентация к родительскому собранию Агрессия детей, ее причины и предупреждение