- Паренхиматозные дистрофии

Содержание
- 2. Типовая форма патологии клетки, которая сопровождается расстройствами метаболизма и приводит к нарушению структуры и функции клеток
- 3. Расстройства регуляции жизнедеятельности клетки (патология рецепторов, нарушение синтеза БАВ), ферментопатии, энергодефицит Расстройства эндокринной регуляции Расстройства нервной
- 4. Инфильтрация – избыточное проникновение продуктов обмена из крови и лимфы в клетку и межклеточное вещество с
- 5. Декомпозиция – распад ультраструктур клеток и межклеточного вещества (мембраны, соединительная ткань) Извращенный синтез – синтез веществ,
- 6. Ультраструктурный – повреждение ядра и органелл Клеточный (атипичные клетки) Тканевый (доброкачественные опухоли) Огранный (жировая дистрофия миокарда)
- 7. По преобладанию морфологических изменений в паренхиме или строме – паренхиматозные, стромально-сосудистрые (мехенхимальные, интерстициальные), смешанные По преобладанию
- 8. В зависимости от влияния генетических факторов – наследственные и приобретенные По распространенности – местные и общие
- 9. Проявления нарушения обмена веществ в высокоспециализированных тканях По виду обмена веществ – белковые, жировые, углеводные Паренхиматозные
- 10. 1. паренхиматозные белковые дистрофии - диспротеинозы
- 11. Морфогенез белковых дистрофий Денатурация и коагуляция белков цитоплазмы Гиалиново-капельная дистрофия Коагуляционный фокальный некроз Коакуляционный тотальный некроз
- 12. Зернистая – гиперплазия органелл (мутное набухание) Гиалиново-капельная – крупные белковые капли, сливающиеся между собой, деструкция органелл
- 13. Фенилкетонурия – дефицит фенил-аланин-4-гидроксилазы (олигофрения) Тирозиноз – дефицит тирозинаминострансферазы (тубулопатии) Алкаптоурия (охроноз) Наследственные белковые дистрофии
- 14. Аутосомно-рецессивное заболевание, в основе которого лежит недостаточность оксидазы гомогентизиновой кислоты, что блокирует метаболизм фенилаланина и тирозина
- 15. Развивается артропатия – эрозии и изнашивание хрящей (поражаются межпозвонковые диски, коленные, плечевые, бедренные суставы) Моча черная
- 16. Виды липидов Сложные (липопротеины, фосфолипиды) – мембраны клеток Прострые – эфиры холестерина и жирных кислот (нейтральные
- 17. Морфологический эквивалент сердечной недостаточности В основе – деструкция митохондрий, что приводит к нарушению окисления жирных кислот
- 18. Отложение капель жира в эпителии проксимальных и дистальных канальцев (почки увеличены, дряблые, корковое вещество набухшее) –
- 19. Пылевидное, мелкокапельное и крупнокапельное ожирение, печень увеличена в размерах, дряблая, желто-коричневая («гусиная печень») Жировая дистрофия печени
- 20. Гипоксия (нарушение окисления ЖК) Инфекции (дифтерия, туберкулез, сепсис) – блокада окислительных ферментов Интоксикации (фосфор, мышьяк, хлороформ,
- 21. Наследственные паренхиматозные жировые дистрофии
- 22. Это наиболее частая форма ганглиозидозов Gm2, связана с мутацией, поражающей локус α-субъединицы на хромосоме 15 и
- 23. Морфогенез Ганглиозиды Gm2 накапливаются преимущественно в нейронах ЦНС и ВНС, а также в сетчатке глаза Под
- 24. С 6-месячного возраста у детей прогрессируют двигательные и психические нарушения (расстройства координации, вялые параличи, снижение остроты
- 25. Отложение в лизосомах сфингомиелина (липиды нервной ткани) и холестерина, связанное с недостаточностью сфингомиелиназы При микроскопии –
- 26. На сетчатке – вишнево-красные пятна (как при болезни Тэя-Сакса) Увеличение живота за счет селезенки Образование ксантом
- 27. Аутосомно-рецессивное заболевание, связанное с мутацией локуса глюкоцереброзидазы в позиции 1q21 Пораженный ген кодирует указанный фермент, который
- 28. 1 тип (отсутствует поражение нейронов) – глюкоцереброзиды накапливаются в фагоцитах, поражается селезенка и скелет (доброкачественная форма)
- 29. Микроскопически определяют наличие клеток Гоше – раздутые макрофаги в печени, селезенке, костном мозге, л/у, легких, при
- 30. В головном мозге клетки Гоше скапливаются в периваскулярных пространствах вокруг нейронов (нейроны сморщены, подвергаются некрозу, что
- 31. Пересадка костного мозга (основной дефект локализуется в мононуклеарных фагоцитах) Перенос гена цереброзидазы в клетки больного Создана
- 32. 3 вида углеводов Гликозаминогликаны Полисахариды (гликоген) Гликопротеиды (муцин – слизь, мукоиды – в тканях) 3. Паренхиматозные
- 33. Нейтральные (прочно связаны с белками) Кислые (непрочно связаны с метаболитами, осуществляют их транспорт) – гиалуроновая кислота,
- 34. Одна из форм болезней лизосомального накопления Группа заболеваний, развивающихся в результате генетически обусловленного недостатка специфических лизосомальных
- 35. Ферменты, участвующие в расщеплении этих молекул, отрезают терминальные сахарные группы от полисахаридных цепей, расположенных вдоль полипептида
- 36. Известно 7 вариантов МПС Все наследуются как аутосомно-рецессивные заболевания, кроме синдрома Пфаундлера-Гурлер (или синдрома Хантера) –
- 37. Прогрессирующее заболевание с поражением многих органов – печени, селезенки, сердца, сосудов Характерны скопления МПС в макрофагах,
- 38. Гепатомегалия, спленомегалия Грубые черты лица, помутнение роговицы, тугоподвижность суставов, умственная отсталость Клиническая картина МПС
- 39. МПС 1 типа – в основе лежит недостаточность α-L-идуронидазы Пораженные дети нормальны при рождении, в интервале
- 40. Лабильный гликоген (печень, мышцы) Стабильный гликоген (нервная система, сердце, хрящи, сосуды) Гормоны, регулирующие обмен гликогена –
- 41. Печень (снижение запасов гликогена, инфильтрация жирами, включение гликогена в ядрах – «пустые ядра» Глюкозурия – гликогенная
- 42. гликогенозы – без нарушения структуры гликогена
- 43. Гликогенозы – с изменением структуры гликогена
- 44. Дефицит глюкозо-6-фосфатазы приводит к нарушению превращения глюкозо-6-фосфата в глюкозу Глюкозо-6-фосфат стимулирует синтез глкогена, липидов с избыточным
- 45. Гепатомегалия – печень увеличена, при биопсии выявляют увеличение гепатоцитов и накопление в их цитоплазме гликогена, появление
- 46. Дефицит в печени и мышцах амило-1,6-глюкозидазы (наиболее частый) Основные признаки – гипогликемия и гепатомегалия Наблюдается миопатия,
- 47. Недостаточность фосфорилазы в мышцах Основной симптом – боли и судороги в мышцах после физической нагрузке, появление
- 48. Обусловлен недостаточностью α-глюкозидазы в лизосомах и является примером болезни накопления Характерно поражение скелетных мышц (слабость и
- 49. В печени откладывается аномальный полисахарид, сходный с амилопектином (недостаточность 1,4-глюкан,6-α-глюкозилтрансферазы У детей развивается гепатомегалия, гипотония мышц,
- 50. Наследственный аутосомно-рецессивный дефицит гликогенсинтетазы В тканях у больных остсутствуют отложения гликогена У детей – утренняя гипогликемия,
- 51. Слизистая дистрофия – накопление муцинов или мукоидов (слизеподобные вещества) Коллоидный зоб (гипотиреоз) – отложение муцинов в
- 53. Скачать презентацию

Типовая форма патологии клетки, которая сопровождается расстройствами метаболизма и приводит к
Типовая форма патологии клетки, которая сопровождается расстройствами метаболизма и приводит к

Расстройства регуляции жизнедеятельности клетки (патология рецепторов, нарушение синтеза БАВ), ферментопатии, энергодефицит
Расстройства
Расстройства регуляции жизнедеятельности клетки (патология рецепторов, нарушение синтеза БАВ), ферментопатии, энергодефицит
Расстройства

Инфильтрация – избыточное проникновение продуктов обмена из крови и лимфы в
Инфильтрация – избыточное проникновение продуктов обмена из крови и лимфы в

Декомпозиция – распад ультраструктур клеток и межклеточного вещества (мембраны, соединительная ткань)
Извращенный
Декомпозиция – распад ультраструктур клеток и межклеточного вещества (мембраны, соединительная ткань)
Извращенный

Ультраструктурный – повреждение ядра и органелл
Клеточный (атипичные клетки)
Тканевый (доброкачественные опухоли)
Огранный (жировая
Ультраструктурный – повреждение ядра и органелл
Клеточный (атипичные клетки)
Тканевый (доброкачественные опухоли)
Огранный (жировая

По преобладанию морфологических изменений в паренхиме или строме – паренхиматозные, стромально-сосудистрые
По преобладанию морфологических изменений в паренхиме или строме – паренхиматозные, стромально-сосудистрые

В зависимости от влияния генетических факторов – наследственные и приобретенные
По распространенности
В зависимости от влияния генетических факторов – наследственные и приобретенные
По распространенности

Проявления нарушения обмена веществ в высокоспециализированных тканях
По виду обмена веществ –
Проявления нарушения обмена веществ в высокоспециализированных тканях
По виду обмена веществ –

1. паренхиматозные белковые дистрофии - диспротеинозы
1. паренхиматозные белковые дистрофии - диспротеинозы

Морфогенез белковых дистрофий
Денатурация и коагуляция белков цитоплазмы
Гиалиново-капельная дистрофия
Коагуляционный фокальный некроз
Коакуляционный тотальный
Морфогенез белковых дистрофий
Денатурация и коагуляция белков цитоплазмы
Гиалиново-капельная дистрофия
Коагуляционный фокальный некроз
Коакуляционный тотальный

Зернистая – гиперплазия органелл (мутное набухание)
Гиалиново-капельная – крупные белковые капли, сливающиеся
Зернистая – гиперплазия органелл (мутное набухание)
Гиалиново-капельная – крупные белковые капли, сливающиеся

Фенилкетонурия – дефицит фенил-аланин-4-гидроксилазы (олигофрения)
Тирозиноз – дефицит тирозинаминострансферазы (тубулопатии)
Алкаптоурия (охроноз)
Наследственные белковые
Фенилкетонурия – дефицит фенил-аланин-4-гидроксилазы (олигофрения)
Тирозиноз – дефицит тирозинаминострансферазы (тубулопатии)
Алкаптоурия (охроноз)
Наследственные белковые

Аутосомно-рецессивное заболевание, в основе которого лежит недостаточность оксидазы гомогентизиновой кислоты, что
Аутосомно-рецессивное заболевание, в основе которого лежит недостаточность оксидазы гомогентизиновой кислоты, что

Развивается артропатия – эрозии и изнашивание хрящей (поражаются межпозвонковые диски, коленные,
Развивается артропатия – эрозии и изнашивание хрящей (поражаются межпозвонковые диски, коленные,

Виды липидов
Сложные (липопротеины, фосфолипиды) – мембраны клеток
Прострые – эфиры холестерина и
Виды липидов
Сложные (липопротеины, фосфолипиды) – мембраны клеток
Прострые – эфиры холестерина и

Морфологический эквивалент сердечной недостаточности
В основе – деструкция митохондрий, что приводит к
Морфологический эквивалент сердечной недостаточности
В основе – деструкция митохондрий, что приводит к

Отложение капель жира в эпителии проксимальных и дистальных канальцев (почки увеличены,
Отложение капель жира в эпителии проксимальных и дистальных канальцев (почки увеличены,

Пылевидное, мелкокапельное и крупнокапельное ожирение, печень увеличена в размерах, дряблая, желто-коричневая
Пылевидное, мелкокапельное и крупнокапельное ожирение, печень увеличена в размерах, дряблая, желто-коричневая

Гипоксия (нарушение окисления ЖК)
Инфекции (дифтерия, туберкулез, сепсис) – блокада окислительных ферментов
Интоксикации
Гипоксия (нарушение окисления ЖК)
Инфекции (дифтерия, туберкулез, сепсис) – блокада окислительных ферментов
Интоксикации
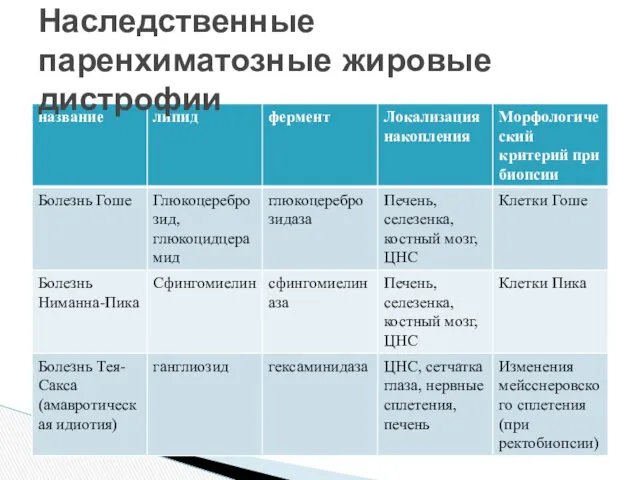
Наследственные паренхиматозные жировые дистрофии
Наследственные паренхиматозные жировые дистрофии

Это наиболее частая форма ганглиозидозов Gm2, связана с мутацией, поражающей локус
Это наиболее частая форма ганглиозидозов Gm2, связана с мутацией, поражающей локус

Морфогенез
Ганглиозиды Gm2 накапливаются преимущественно в нейронах ЦНС и ВНС, а
Морфогенез
Ганглиозиды Gm2 накапливаются преимущественно в нейронах ЦНС и ВНС, а

С 6-месячного возраста у детей прогрессируют двигательные и психические нарушения (расстройства
С 6-месячного возраста у детей прогрессируют двигательные и психические нарушения (расстройства

Отложение в лизосомах сфингомиелина (липиды нервной ткани) и холестерина, связанное с
Отложение в лизосомах сфингомиелина (липиды нервной ткани) и холестерина, связанное с

На сетчатке – вишнево-красные пятна (как при болезни Тэя-Сакса)
Увеличение живота за
На сетчатке – вишнево-красные пятна (как при болезни Тэя-Сакса)
Увеличение живота за

Аутосомно-рецессивное заболевание, связанное с мутацией локуса глюкоцереброзидазы в позиции 1q21
Пораженный
Аутосомно-рецессивное заболевание, связанное с мутацией локуса глюкоцереброзидазы в позиции 1q21
Пораженный

1 тип (отсутствует поражение нейронов) – глюкоцереброзиды накапливаются в фагоцитах, поражается
1 тип (отсутствует поражение нейронов) – глюкоцереброзиды накапливаются в фагоцитах, поражается

Микроскопически определяют наличие клеток Гоше – раздутые макрофаги в печени, селезенке,
Микроскопически определяют наличие клеток Гоше – раздутые макрофаги в печени, селезенке,

В головном мозге клетки Гоше скапливаются в периваскулярных пространствах вокруг нейронов
В головном мозге клетки Гоше скапливаются в периваскулярных пространствах вокруг нейронов

Пересадка костного мозга (основной дефект локализуется в мононуклеарных фагоцитах)
Перенос гена цереброзидазы
Пересадка костного мозга (основной дефект локализуется в мононуклеарных фагоцитах)
Перенос гена цереброзидазы

3 вида углеводов
Гликозаминогликаны
Полисахариды (гликоген)
Гликопротеиды (муцин – слизь, мукоиды – в тканях)
3.
3 вида углеводов
Гликозаминогликаны
Полисахариды (гликоген)
Гликопротеиды (муцин – слизь, мукоиды – в тканях)
3.

Нейтральные (прочно связаны с белками)
Кислые (непрочно связаны с метаболитами, осуществляют их
Нейтральные (прочно связаны с белками)
Кислые (непрочно связаны с метаболитами, осуществляют их

Одна из форм болезней лизосомального накопления
Группа заболеваний, развивающихся в результате генетически
Одна из форм болезней лизосомального накопления
Группа заболеваний, развивающихся в результате генетически

Ферменты, участвующие в расщеплении этих молекул, отрезают терминальные сахарные группы от
Ферменты, участвующие в расщеплении этих молекул, отрезают терминальные сахарные группы от

Известно 7 вариантов МПС
Все наследуются как аутосомно-рецессивные заболевания, кроме синдрома Пфаундлера-Гурлер
Известно 7 вариантов МПС
Все наследуются как аутосомно-рецессивные заболевания, кроме синдрома Пфаундлера-Гурлер

Прогрессирующее заболевание с поражением многих органов – печени, селезенки, сердца, сосудов
Характерны
Прогрессирующее заболевание с поражением многих органов – печени, селезенки, сердца, сосудов
Характерны

Гепатомегалия, спленомегалия
Грубые черты лица, помутнение роговицы, тугоподвижность суставов, умственная отсталость
Клиническая картина
Гепатомегалия, спленомегалия
Грубые черты лица, помутнение роговицы, тугоподвижность суставов, умственная отсталость
Клиническая картина

МПС 1 типа – в основе лежит недостаточность α-L-идуронидазы
Пораженные дети нормальны
МПС 1 типа – в основе лежит недостаточность α-L-идуронидазы
Пораженные дети нормальны

Лабильный гликоген (печень, мышцы)
Стабильный гликоген (нервная система, сердце, хрящи, сосуды)
Гормоны, регулирующие
Лабильный гликоген (печень, мышцы)
Стабильный гликоген (нервная система, сердце, хрящи, сосуды)
Гормоны, регулирующие

Печень (снижение запасов гликогена, инфильтрация жирами, включение гликогена в ядрах –
Печень (снижение запасов гликогена, инфильтрация жирами, включение гликогена в ядрах –

гликогенозы – без нарушения структуры гликогена
гликогенозы – без нарушения структуры гликогена

Гликогенозы – с изменением структуры гликогена
Гликогенозы – с изменением структуры гликогена

Дефицит глюкозо-6-фосфатазы приводит к нарушению превращения глюкозо-6-фосфата в глюкозу
Глюкозо-6-фосфат стимулирует синтез
Дефицит глюкозо-6-фосфатазы приводит к нарушению превращения глюкозо-6-фосфата в глюкозу
Глюкозо-6-фосфат стимулирует синтез

Гепатомегалия – печень увеличена, при биопсии выявляют увеличение гепатоцитов и накопление
Гепатомегалия – печень увеличена, при биопсии выявляют увеличение гепатоцитов и накопление

Дефицит в печени и мышцах амило-1,6-глюкозидазы (наиболее частый)
Основные признаки – гипогликемия
Дефицит в печени и мышцах амило-1,6-глюкозидазы (наиболее частый)
Основные признаки – гипогликемия

Недостаточность фосфорилазы в мышцах
Основной симптом – боли и судороги в мышцах
Недостаточность фосфорилазы в мышцах
Основной симптом – боли и судороги в мышцах

Обусловлен недостаточностью α-глюкозидазы в лизосомах и является примером болезни накопления
Характерно поражение
Обусловлен недостаточностью α-глюкозидазы в лизосомах и является примером болезни накопления
Характерно поражение

В печени откладывается аномальный полисахарид, сходный с амилопектином (недостаточность 1,4-глюкан,6-α-глюкозилтрансферазы
У детей
В печени откладывается аномальный полисахарид, сходный с амилопектином (недостаточность 1,4-глюкан,6-α-глюкозилтрансферазы
У детей

Наследственный аутосомно-рецессивный дефицит гликогенсинтетазы
В тканях у больных остсутствуют отложения гликогена
У детей
Наследственный аутосомно-рецессивный дефицит гликогенсинтетазы
В тканях у больных остсутствуют отложения гликогена
У детей

Слизистая дистрофия – накопление муцинов или мукоидов (слизеподобные вещества)
Коллоидный зоб (гипотиреоз)
Слизистая дистрофия – накопление муцинов или мукоидов (слизеподобные вещества)
Коллоидный зоб (гипотиреоз)
 Форум профессионалов молодёжной сферы #смородина
Форум профессионалов молодёжной сферы #смородина Переливание крови
Переливание крови Аргентина. Визитная карточка
Аргентина. Визитная карточка Подготовка объектов автоматизации к внедрению и опытной эксплуатации
Подготовка объектов автоматизации к внедрению и опытной эксплуатации Железобетонные бункера
Железобетонные бункера Проект Прошлое и настоящее усадьбы Ланских-Араповых
Проект Прошлое и настоящее усадьбы Ланских-Араповых Родина ретровірусів. Віруси – збудники СНІДу та онкогенних інфекцій
Родина ретровірусів. Віруси – збудники СНІДу та онкогенних інфекцій Лингвистика как наука о языке. Общее понятие о языке. Язык как знаковая система
Лингвистика как наука о языке. Общее понятие о языке. Язык как знаковая система Роль женщин в истории истории математики
Роль женщин в истории истории математики Преступления против государственной власти, интересов государственной службы и службы в органах местного самоуправления
Преступления против государственной власти, интересов государственной службы и службы в органах местного самоуправления Луна
Луна День Матери 2015 год
День Матери 2015 год Самое-самое интересное о железной дороге
Самое-самое интересное о железной дороге Пропорции головы человека (ИЗО)
Пропорции головы человека (ИЗО) Общая характеристика грибов
Общая характеристика грибов Выявление особенностей связного повествовательного высказывания у старших дошкольников с интеллектуальными нарушениями
Выявление особенностей связного повествовательного высказывания у старших дошкольников с интеллектуальными нарушениями Семейный праздник 23 + 8 (2 класс)
Семейный праздник 23 + 8 (2 класс) Этапы развития искусства Ренессанса
Этапы развития искусства Ренессанса Конфуцианство. Жизнь Конфуция и его учения
Конфуцианство. Жизнь Конфуция и его учения Детям о космосе
Детям о космосе Программная инженерия. Тестирование
Программная инженерия. Тестирование Игра Третий лишний
Игра Третий лишний Векторы на плоскости
Векторы на плоскости ЛРС седативного действия
ЛРС седативного действия Части речи. Существительное
Части речи. Существительное Автоматизация процесса извлечения метаданных из слабоструктурированных документов
Автоматизация процесса извлечения метаданных из слабоструктурированных документов Социальная политика государства
Социальная политика государства моя визитка
моя визитка