- Воспроизведенные и гибридные лекарственные средства. Подготовка регистрационного досье

Содержание
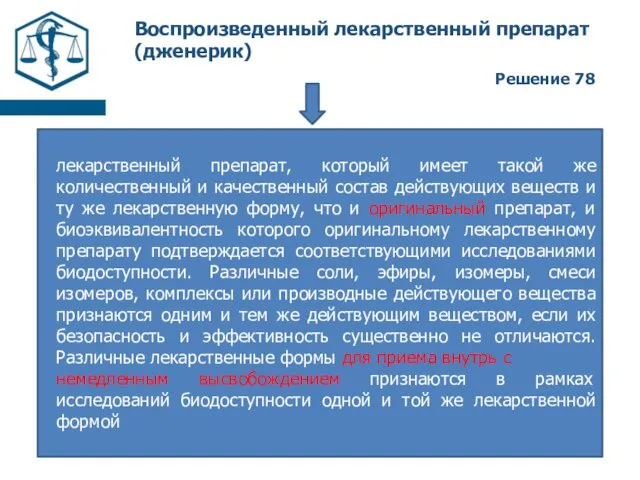
- 2. Воспроизведенный лекарственный препарат (дженерик) Решение 78 лекарственный препарат, который имеет такой же количественный и качественный состав
- 3. Воспроизведенный лекарственный препарат (дженерик) Решение 85 лекарственный препарат, имеющий такой же качественный и количественный состав действующих
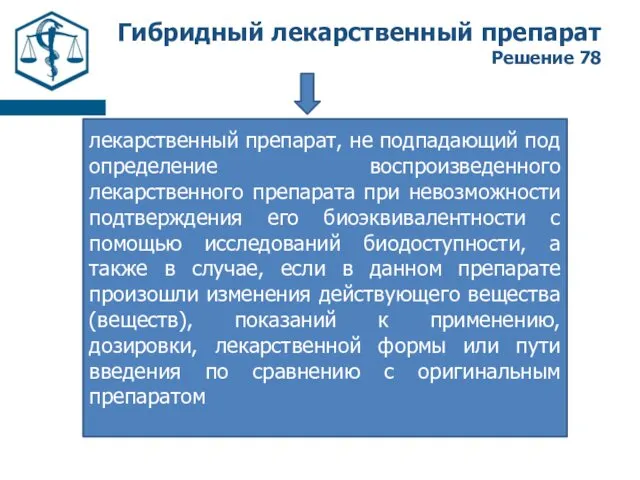
- 4. Гибридный лекарственный препарат Решение 78 лекарственный препарат, не подпадающий под определение воспроизведенного лекарственного препарата при невозможности
- 5. Гибридный лекарственный препарат Решение 85 лекарственный препарат, не подпадающий под определение воспроизведенного лекарственного препарата, приведенного в
- 6. Оригинальный лекарственный препарат Решение 78, 85 лекарственный препарат с новым действующим веществом, который был первым зарегистрирован
- 7. Референтный лекарственный препарат Решение 78, 85 лекарственный препарат, который используется в качестве препарата сравнения и является
- 8. Требования к представлению документов регламентируют: Решение Евразийской экономической комиссии №78 от 03.11.2016 г. «О правилах регистрации
- 9. Требования к представлению документов регламентируют Приложения 1-5: Требования к документам регистрационного досье (в формате ОТД) –
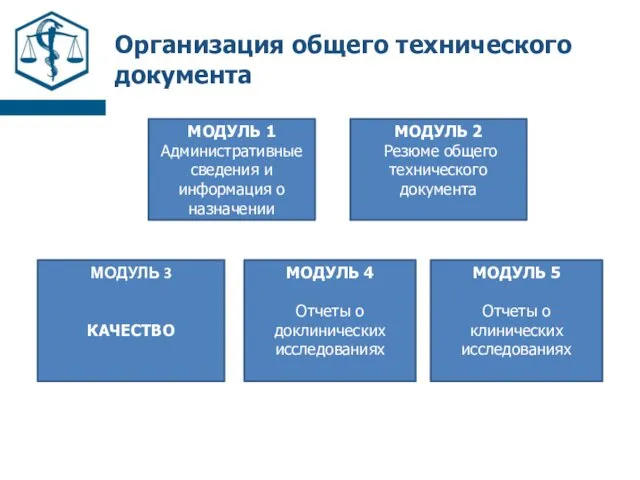
- 11. Организация общего технического документа МОДУЛЬ 1 Административные сведения и информация о назначении МОДУЛЬ 2 Резюме общего
- 12. Организация общего технического документа
- 13. Регистрация воспроизведенного лекарственного препарата Последовательно Процедура взаимного признания Референтное государство (рынок государства) В государствах признания (по
- 14. Регистрация и экспертиза лекарственного препарата в референтном государстве заявление на бумажном носителе и (или) в виде
- 15. Экспертиза лекарственного препарата в референтном государстве а) оценку полноты, комплектности и правильности оформления документов, представленных в
- 16. Рассмотрение материалов Запрос (90 дней, общий срок запросов не более 180 календарных дней) - не входит
- 17. Положительное решение регистрационное удостоверение лекарственного препарата по форме согласно приложению № 17 инструкцию по медицинскому применению
- 18. По результатам экспертизы заключительный экспертный отчет по форме согласно приложению № 16 к настоящим Правилам Экспертные
- 19. Отказ а) соотношение ожидаемой пользы к возможным рискам, связанным с применением лекарственного препарата, не является благоприятным;
- 20. Регистрация и экспертиза лекарственного препарата по процедуре взаимного признания в государстве (государствах) признания заявления на регистрацию
- 21. Уполномоченный орган (экспертная организация) референтного государства доступ для уполномоченных органов (экспертных организаций) государств признания к регистрационному
- 22. Порядок регистрации и экспертизы по децентрализованной процедуре в референтном государстве и государствах признания Выбор референтного государства
- 23. Референтное государство заявление о регистрации лекарственного препарата по установленной форме на бумажном носителе и (или) в
- 24. Государства признания (не позднее 14 рабочих дней) заявление на бумажном носителе и (или) в виде электронного
- 25. Экспертиза Референтное Досье Документы и данные представленных заявителем, на предмет безопасности, эффективности и качества проведение лабораторных
- 26. Рассмотрение материалов Запрос (90 дней, общий срок запросов не более 180 календарных дней) - не входит
- 27. Процедура приведения регистрационного досье лекарственного препарата, зарегистрированного до вступления в силу Соглашения о единых принципах и
- 28. Необходимо представить: письменное подтверждение, что документы и данные, содержащиеся в представленном обновленном регистрационном досье в формате
- 29. Референтное государство (факт регистрации) заявление по установленной форме на бумажном носителе и (или) в виде электронного
- 30. Рассмотрение материалов Запрос (90 дней, общий срок запросов не более 180 календарных дней) - не входит
- 31. Процедура взаимного признания в государствах-членах, в которых данный лекарственный препарат не был зарегистрирован (после приведения в
- 32. Процедура приведения в соответствие Все имеющиеся данные доклинических и клинических исследований, выполненных до вступления в силу
- 33. В случае необходимости подготовки экспертного отчета по оценке для проведения процедуры взаимного признания в государстве, в
- 34. МОДУЛЬ 1 Административные сведения и информация о назначении 1.0. Сопроводительное письмо 1.1. Содержание досье, необходимо представить
- 35. Общая характеристика лекарственного препарата, инструкция по медицинскому применению, маркировка Проекты ОХЛП, ИМП (на русском языке) Макеты
- 36. Пользовательское тестирование При представлении результатов пользовательского тестирования необходимо кратко обобщить, как было проведено тестирование и каким
- 37. 1.3 ОХЛП и ИМП Общая характеристика лекарственного препарата и инструкция по медицинскому применению воспроизведенного лекарственного препарата
- 38. В случае отличия показаний к применению в сторону расширения или режима дозирования либо пути введения в
- 39. 1.4.Информация по регуляторному статусу лекарственного препарата в других странах (при наличии). Перечень стран, в которых лекарственный
- 40. 1.7. Информация о специалистах Информация о специалисте, подготовившем резюме по качеству. Информация о специалисте, подготовившем резюме
- 41. 1.8 Специфические требования для различных типов заявлений 1.8.1. Письмо держателя регистрационного удостоверения о дополнительном торговом наименовании
- 42. 1.8.2. Документы по клиническим исследованиям (если применимо): Разрешение уполномоченного органа на проведение клинического исследования, в том
- 43. 1.8 Специфические требования для различных типов заявлений/воспроизведенные 1.8.2 Документы по клиническим исследованиям (если применимо) Резюме (до
- 44. 1.8 Специфические требования для различных типов заявлений/гибридные 1.8.2 Документы по клиническим исследованиям Резюме (до 5 страниц)
- 45. Информация относительно фармаконадзора заявителя в государстве-члене. Мастер-файл системы фармаконадзора держателя регистрационного удостоверения представляется в случае, когда
- 46. МОДУЛЬ 2 Резюме общего технического документа Резюме химической и биологической документации, доклинических и клинических данных, представленных
- 47. МОДУЛЬ 2 Резюме общего технического документа 2.1 Сводное содержание ОТД (модули 2 – 5) 2.2 Введение
- 48. 2.4. Доклинический обзор 2.5. Клинический обзор - резюме профиля примесей активного вещества (и в соответствующих случаях
- 49. 2.4. Доклинический обзор 2.5. Клинический обзор - ранее неизвестные или следующие из характеристик препарата и (или)
- 50. 2.6. Резюме по доклиническим исследованиям Резюме доклинических данных нужно представлять на основе фактических результатов фармакологических, фармакокинетических
- 51. 2.7. Резюме клинических данных Необходимо представить подробное с приведением фактических данных резюме клинической информации по изучению
- 52. МОДУЛЬ 3. Качество. (представляется полностью) 3.1. Содержание 3.2. Основные сведения 3.2.S. Активная фармацевтическая субстанция. 3.2.S.1. Общая
- 53. 3.2.P. Лекарственный препарат 3.2.P.1.Описание и состав лекарственного препарата 3.2.P.2. Фармацевтическая разработка 3.2.P.3. Процесс производства лекарственного препарата
- 54. 3.2.А. Дополнения. 3.2.А.1. Производственные помещения и оборудование. 3.2.А.2. Оценка безопасности лекарственных препаратов относительно наличия посторонних агентов.
- 55. МОДУЛЬ 4 Отчеты о доклинических (неклинических) исследованиях 4.1. Содержание модуля 4 4.2. Отчеты об исследованиях 4.2.1.
- 56. Доклинические исследования Доклинические исследования безопасности лекарственных средств, проведенные в государствах, не являющихся членами Союза, рассматриваются в
- 57. 4.2. Отчеты о доклинических (неклинических) исследованиях В отдельных случаях в соответствии с требованиями по исследованию отдельных
- 58. МОДУЛЬ 5 Отчеты о клинических испытаниях 5.1 Содержание модуля 5 5.2 Табличный перечень всех клинических исследований
- 59. 5.3 Отчеты о клинических исследованиях 5.3.1. Отчеты о биофармацевтических исследованиях – должны быть включены результаты исследований
- 60. Отчеты о проведении исследований биоэквивалентности - сведения об исследователе (с указанием его рабочего места), - организации,
- 61. Сведения о референтном препарате торговое наименование; дозировка; лекарственная форма; держатель регистрационного удостоверения; дата регистрации; номер регистрационного
- 62. Сведения о тестируемом препарате наименование состав размер серии дату производства дату окончания срока годности (по возможности)
- 63. К отчету по проведенному исследованию необходимо приложить данные лабораторных и инструментальных методов исследования, Данные статистической обработки
- 64. Следует представить дополнительную информацию профили безопасности и (или) эффективности заявленного лекарственного препарата не отличаются от таковых
- 65. Проведение исследований биоэквивалентности не требуется Лекарственные препараты для приема внутрь (I, III класс по БКС) Растворы
- 66. Дополнительные данные для гибридных ЛП
- 67. Дополнительные данные для гибридных ЛП
- 68. Дополнительные данные для гибридных ЛП
- 69. Экспертиза Оценка полноты, комплектности и правильности оформления документов; Оценка представленных данных по аспектам качества, безопасности и
- 70. По результатам регистрации лекарственного препарата уполномоченный орган каждого государства-члена, зарегистрировавшего лекарственный препарат, выдает регистрационное удостоверение лекарственного
- 71. ПРИЛОЖЕНИЕ № 17 к Правилам регистрации и экспертизы лекарственных средств для медицинского применения ФОРМА регистрационного удостоверения
- 73. Скачать презентацию
Воспроизведенный лекарственный препарат (дженерик)
Решение 78
лекарственный препарат, который имеет такой же
Воспроизведенный лекарственный препарат (дженерик)
Решение 78
лекарственный препарат, который имеет такой же

Воспроизведенный лекарственный препарат (дженерик)
Решение 85
лекарственный препарат, имеющий такой же качественный
Воспроизведенный лекарственный препарат (дженерик)
Решение 85
лекарственный препарат, имеющий такой же качественный
Гибридный лекарственный препарат
Решение 78
лекарственный препарат, не подпадающий под определение воспроизведенного лекарственного
Гибридный лекарственный препарат
Решение 78
лекарственный препарат, не подпадающий под определение воспроизведенного лекарственного

Гибридный лекарственный препарат
Решение 85
лекарственный препарат, не подпадающий под определение воспроизведенного
Гибридный лекарственный препарат
Решение 85
лекарственный препарат, не подпадающий под определение воспроизведенного

Оригинальный лекарственный препарат
Решение 78, 85
лекарственный препарат с новым действующим веществом, который
Оригинальный лекарственный препарат
Решение 78, 85
лекарственный препарат с новым действующим веществом, который

Референтный лекарственный препарат
Решение 78, 85
лекарственный препарат, который используется в качестве
Референтный лекарственный препарат
Решение 78, 85
лекарственный препарат, который используется в качестве

Требования к представлению документов регламентируют:
Решение Евразийской экономической комиссии №78
от 03.11.2016
Требования к представлению документов регламентируют:
Решение Евразийской экономической комиссии №78
от 03.11.2016

Требования к представлению документов регламентируют Приложения 1-5:
Требования к документам регистрационного досье
Требования к представлению документов регламентируют Приложения 1-5:
Требования к документам регистрационного досье
Организация общего технического документа
МОДУЛЬ 1
Административные сведения и информация о назначении
МОДУЛЬ 2
Организация общего технического документа
МОДУЛЬ 1
Административные сведения и информация о назначении
МОДУЛЬ 2
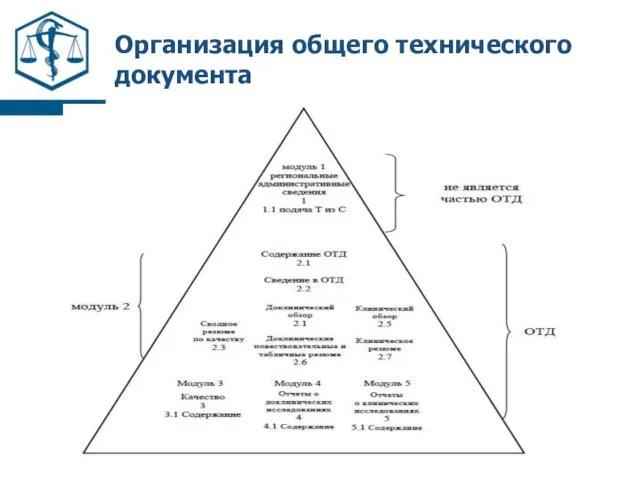
Организация общего технического документа
Организация общего технического документа

Регистрация воспроизведенного лекарственного препарата
Последовательно
Процедура взаимного признания
Референтное государство (рынок государства)
В государствах признания
Регистрация воспроизведенного лекарственного препарата
Последовательно
Процедура взаимного признания
Референтное государство (рынок государства)
В государствах признания

Регистрация и экспертиза лекарственного препарата в референтном государстве
заявление на бумажном носителе
Регистрация и экспертиза лекарственного препарата в референтном государстве
заявление на бумажном носителе

Экспертиза лекарственного препарата в референтном государстве
а) оценку полноты, комплектности и правильности
Экспертиза лекарственного препарата в референтном государстве
а) оценку полноты, комплектности и правильности

Рассмотрение материалов
Запрос (90 дней, общий срок запросов не более 180 календарных
Рассмотрение материалов
Запрос (90 дней, общий срок запросов не более 180 календарных

Положительное решение
регистрационное удостоверение лекарственного препарата по форме согласно приложению № 17
инструкцию
Положительное решение
регистрационное удостоверение лекарственного препарата по форме согласно приложению № 17
инструкцию
По результатам экспертизы
заключительный экспертный отчет по форме согласно приложению № 16
По результатам экспертизы
заключительный экспертный отчет по форме согласно приложению № 16

Отказ
а) соотношение ожидаемой пользы к возможным рискам, связанным с применением
Отказ
а) соотношение ожидаемой пользы к возможным рискам, связанным с применением

Регистрация и экспертиза лекарственного препарата по процедуре взаимного признания в государстве
Регистрация и экспертиза лекарственного препарата по процедуре взаимного признания в государстве
Уполномоченный орган (экспертная организация) референтного государства
доступ для уполномоченных органов (экспертных организаций)
Уполномоченный орган (экспертная организация) референтного государства
доступ для уполномоченных органов (экспертных организаций)

Порядок регистрации и экспертизы по децентрализованной процедуре в референтном государстве и
Порядок регистрации и экспертизы по децентрализованной процедуре в референтном государстве и

Референтное государство
заявление о регистрации лекарственного препарата по установленной форме на бумажном
Референтное государство
заявление о регистрации лекарственного препарата по установленной форме на бумажном

Государства признания (не позднее 14 рабочих дней)
заявление на бумажном носителе и
Государства признания (не позднее 14 рабочих дней)
заявление на бумажном носителе и
Экспертиза
Референтное
Досье
Документы и данные представленных заявителем, на предмет безопасности, эффективности и
Экспертиза
Референтное
Досье
Документы и данные представленных заявителем, на предмет безопасности, эффективности и
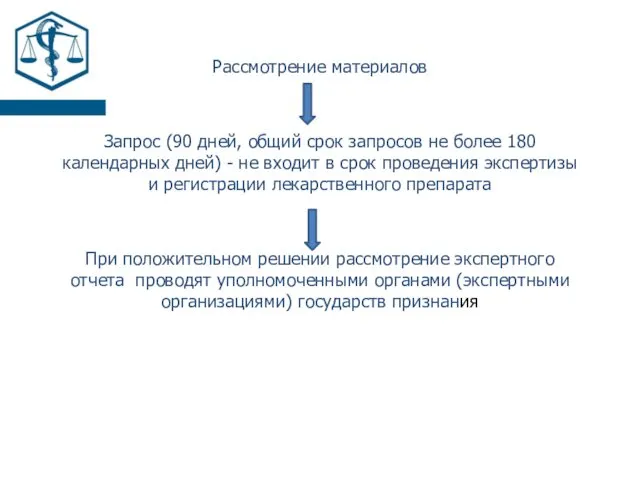
Рассмотрение материалов
Запрос (90 дней, общий срок запросов не более 180 календарных
Рассмотрение материалов
Запрос (90 дней, общий срок запросов не более 180 календарных

Процедура приведения регистрационного досье лекарственного препарата, зарегистрированного до вступления в силу
Процедура приведения регистрационного досье лекарственного препарата, зарегистрированного до вступления в силу

Необходимо представить:
письменное подтверждение, что документы и данные, содержащиеся в представленном
Необходимо представить:
письменное подтверждение, что документы и данные, содержащиеся в представленном

Референтное государство (факт регистрации)
заявление по установленной форме на бумажном носителе и
Референтное государство (факт регистрации)
заявление по установленной форме на бумажном носителе и

Рассмотрение материалов
Запрос (90 дней, общий срок запросов не более 180 календарных
Рассмотрение материалов
Запрос (90 дней, общий срок запросов не более 180 календарных

Процедура взаимного признания в государствах-членах, в которых данный лекарственный препарат не
Процедура взаимного признания в государствах-членах, в которых данный лекарственный препарат не

Процедура приведения в соответствие
Все имеющиеся данные доклинических и клинических исследований,
Процедура приведения в соответствие
Все имеющиеся данные доклинических и клинических исследований,

В случае необходимости подготовки экспертного отчета по оценке для проведения процедуры
В случае необходимости подготовки экспертного отчета по оценке для проведения процедуры

МОДУЛЬ 1
Административные сведения и информация о назначении
1.0. Сопроводительное письмо
1.1. Содержание
МОДУЛЬ 1
Административные сведения и информация о назначении
1.0. Сопроводительное письмо
1.1. Содержание

Общая характеристика лекарственного препарата, инструкция по медицинскому применению, маркировка
Проекты ОХЛП, ИМП
Общая характеристика лекарственного препарата, инструкция по медицинскому применению, маркировка
Проекты ОХЛП, ИМП

Пользовательское тестирование
При представлении результатов пользовательского тестирования необходимо кратко обобщить, как было
Пользовательское тестирование
При представлении результатов пользовательского тестирования необходимо кратко обобщить, как было

1.3 ОХЛП и ИМП
Общая характеристика лекарственного препарата и инструкция по медицинскому
1.3 ОХЛП и ИМП
Общая характеристика лекарственного препарата и инструкция по медицинскому

В случае отличия показаний к применению в сторону расширения или режима
В случае отличия показаний к применению в сторону расширения или режима

1.4.Информация по регуляторному статусу лекарственного препарата в других странах (при наличии).
Перечень
1.4.Информация по регуляторному статусу лекарственного препарата в других странах (при наличии).
Перечень

1.7. Информация о специалистах
Информация о специалисте, подготовившем резюме по качеству.
Информация о
1.7. Информация о специалистах
Информация о специалисте, подготовившем резюме по качеству.
Информация о

1.8 Специфические требования для различных типов заявлений
1.8.1. Письмо держателя регистрационного удостоверения
1.8 Специфические требования для различных типов заявлений
1.8.1. Письмо держателя регистрационного удостоверения

1.8.2. Документы по клиническим исследованиям (если применимо):
Разрешение уполномоченного органа на проведение
1.8.2. Документы по клиническим исследованиям (если применимо):
Разрешение уполномоченного органа на проведение

1.8 Специфические требования для различных типов заявлений/воспроизведенные
1.8.2 Документы по клиническим исследованиям
1.8 Специфические требования для различных типов заявлений/воспроизведенные 1.8.2 Документы по клиническим исследованиям

1.8 Специфические требования для различных типов заявлений/гибридные
1.8.2 Документы по клиническим исследованиям
Резюме
1.8 Специфические требования для различных типов заявлений/гибридные
1.8.2 Документы по клиническим исследованиям
Резюме

Информация относительно фармаконадзора заявителя в государстве-члене.
Мастер-файл системы фармаконадзора держателя регистрационного удостоверения
Информация относительно фармаконадзора заявителя в государстве-члене.
Мастер-файл системы фармаконадзора держателя регистрационного удостоверения

МОДУЛЬ 2
Резюме общего технического документа
Резюме химической и биологической документации, доклинических и
МОДУЛЬ 2
Резюме общего технического документа
Резюме химической и биологической документации, доклинических и

МОДУЛЬ 2
Резюме общего технического документа
2.1 Сводное содержание ОТД (модули 2 –
МОДУЛЬ 2
Резюме общего технического документа
2.1 Сводное содержание ОТД (модули 2 –

2.4. Доклинический обзор
2.5. Клинический обзор
- резюме профиля примесей активного вещества (и
2.4. Доклинический обзор
2.5. Клинический обзор
- резюме профиля примесей активного вещества (и

2.4. Доклинический обзор
2.5. Клинический обзор
- ранее неизвестные или следующие из характеристик
2.4. Доклинический обзор
2.5. Клинический обзор
- ранее неизвестные или следующие из характеристик

2.6. Резюме по доклиническим исследованиям
Резюме доклинических данных нужно представлять на основе
2.6. Резюме по доклиническим исследованиям
Резюме доклинических данных нужно представлять на основе

2.7. Резюме клинических данных
Необходимо представить подробное с приведением фактических данных резюме
2.7. Резюме клинических данных
Необходимо представить подробное с приведением фактических данных резюме

МОДУЛЬ 3. Качество.
(представляется полностью)
3.1. Содержание
3.2. Основные сведения
3.2.S. Активная фармацевтическая субстанция.
3.2.S.1. Общая
МОДУЛЬ 3. Качество.
(представляется полностью)
3.1. Содержание
3.2. Основные сведения
3.2.S. Активная фармацевтическая субстанция.
3.2.S.1. Общая

3.2.P. Лекарственный препарат
3.2.P.1.Описание и состав лекарственного препарата
3.2.P.2. Фармацевтическая разработка
3.2.P.3. Процесс производства
3.2.P. Лекарственный препарат
3.2.P.1.Описание и состав лекарственного препарата
3.2.P.2. Фармацевтическая разработка
3.2.P.3. Процесс производства

3.2.А. Дополнения.
3.2.А.1. Производственные помещения и оборудование.
3.2.А.2. Оценка безопасности лекарственных
3.2.А. Дополнения.
3.2.А.1. Производственные помещения и оборудование.
3.2.А.2. Оценка безопасности лекарственных

МОДУЛЬ 4
Отчеты о доклинических (неклинических) исследованиях
4.1. Содержание модуля 4
4.2. Отчеты
МОДУЛЬ 4
Отчеты о доклинических (неклинических) исследованиях
4.1. Содержание модуля 4
4.2. Отчеты

Доклинические исследования
Доклинические исследования безопасности лекарственных средств, проведенные в государствах, не являющихся
Доклинические исследования
Доклинические исследования безопасности лекарственных средств, проведенные в государствах, не являющихся

4.2. Отчеты о доклинических (неклинических) исследованиях
В отдельных случаях в соответствии с
4.2. Отчеты о доклинических (неклинических) исследованиях
В отдельных случаях в соответствии с

МОДУЛЬ 5
Отчеты о клинических испытаниях
5.1 Содержание модуля 5
5.2 Табличный перечень
МОДУЛЬ 5
Отчеты о клинических испытаниях
5.1 Содержание модуля 5
5.2 Табличный перечень

5.3 Отчеты о клинических исследованиях
5.3.1. Отчеты о биофармацевтических исследованиях – должны
5.3 Отчеты о клинических исследованиях
5.3.1. Отчеты о биофармацевтических исследованиях – должны

Отчеты о проведении исследований биоэквивалентности
- сведения об исследователе (с указанием его
Отчеты о проведении исследований биоэквивалентности
- сведения об исследователе (с указанием его

Сведения о референтном препарате
торговое наименование;
дозировка;
лекарственная форма;
держатель регистрационного удостоверения;
Сведения о референтном препарате
торговое наименование;
дозировка;
лекарственная форма;
держатель регистрационного удостоверения;

Сведения о тестируемом препарате
наименование
состав
размер серии
дату производства
дату окончания срока
Сведения о тестируемом препарате
наименование
состав
размер серии
дату производства
дату окончания срока

К отчету по проведенному исследованию необходимо приложить
данные лабораторных и инструментальных методов
К отчету по проведенному исследованию необходимо приложить
данные лабораторных и инструментальных методов

Следует представить дополнительную информацию профили безопасности и (или) эффективности заявленного лекарственного
Следует представить дополнительную информацию профили безопасности и (или) эффективности заявленного лекарственного

Проведение исследований биоэквивалентности не требуется
Лекарственные препараты для приема внутрь (I, III
Проведение исследований биоэквивалентности не требуется
Лекарственные препараты для приема внутрь (I, III

Дополнительные данные для гибридных ЛП
Дополнительные данные для гибридных ЛП
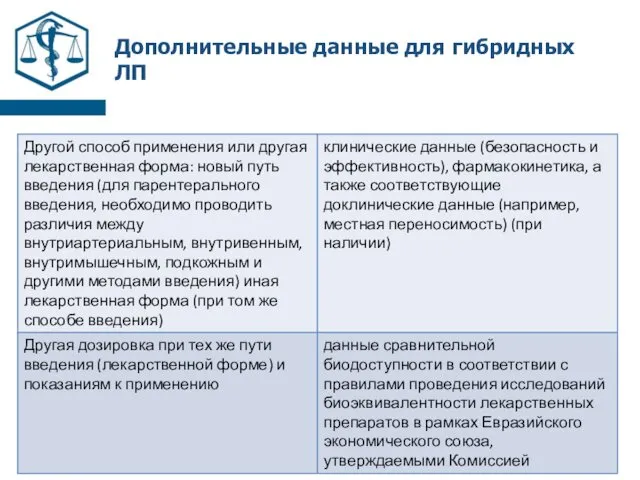
Дополнительные данные для гибридных ЛП
Дополнительные данные для гибридных ЛП

Дополнительные данные для гибридных ЛП
Дополнительные данные для гибридных ЛП

Экспертиза
Оценка полноты, комплектности и правильности оформления документов;
Оценка представленных данных по аспектам
Экспертиза
Оценка полноты, комплектности и правильности оформления документов;
Оценка представленных данных по аспектам

По результатам регистрации лекарственного препарата уполномоченный орган каждого государства-члена, зарегистрировавшего лекарственный
По результатам регистрации лекарственного препарата уполномоченный орган каждого государства-члена, зарегистрировавшего лекарственный

ПРИЛОЖЕНИЕ № 17
к Правилам регистрации и экспертизы
лекарственных средств для
медицинского
ПРИЛОЖЕНИЕ № 17 к Правилам регистрации и экспертизы лекарственных средств для медицинского
 Сойыс малдары. Сойыс малының жалпы сипаттамасы
Сойыс малдары. Сойыс малының жалпы сипаттамасы Исход
Исход Махатма Ганди — один из руководителей и идеолог национальноосвободительного движения Индии
Махатма Ганди — один из руководителей и идеолог национальноосвободительного движения Индии Прессовые формовочные машины
Прессовые формовочные машины Презентация Периоды речевого развития детей
Презентация Периоды речевого развития детей Предельные и непредельные углеводороды. Галоид-, азотсодержащие соединения
Предельные и непредельные углеводороды. Галоид-, азотсодержащие соединения КБА-171
КБА-171 Legal Regime and Protection of Information Technology Products
Legal Regime and Protection of Information Technology Products Презентации Диск
Презентации Диск Классификация камер сгорания
Классификация камер сгорания Библия – книга книг
Библия – книга книг Решение олимпиадных задач по математике.
Решение олимпиадных задач по математике. Применение производной к исследованию функций
Применение производной к исследованию функций Необычные растения и животные в природе.
Необычные растения и животные в природе. Своеобразие органического мира Австралии
Своеобразие органического мира Австралии Житие
Житие Основные понятия и терминология документационного обеспечения систем управления
Основные понятия и терминология документационного обеспечения систем управления Презентация Методическая работа в нашем центре
Презентация Методическая работа в нашем центре Праздничные концерты для любимых женщин. Фотоальбом
Праздничные концерты для любимых женщин. Фотоальбом Экологическая оценка: основные понятия и принципы
Экологическая оценка: основные понятия и принципы Пропедевтика. Эхокардиография (ЭХОКГ С ДКГ)
Пропедевтика. Эхокардиография (ЭХОКГ С ДКГ) Основные направления международного сотрудничества России в области безопасности жизнедеятельности
Основные направления международного сотрудничества России в области безопасности жизнедеятельности Применение ИКТ в работе с родителями детей с нарушениями речи или логопедическая помощь родителям в новом формате
Применение ИКТ в работе с родителями детей с нарушениями речи или логопедическая помощь родителям в новом формате Рациональное строительство с использованием б/у ж/б конструкций
Рациональное строительство с использованием б/у ж/б конструкций ВИЧ
ВИЧ Пілларс
Пілларс 1_Введение
1_Введение ООценка кинических рекомендаций по невропатии лицевого нерва 2013 года с использованием инструмента agree. Область применения
ООценка кинических рекомендаций по невропатии лицевого нерва 2013 года с использованием инструмента agree. Область применения