Наследственные болезни и синдромы. Моногенные заболевания. Частые моногенные заболевания (муковисцидоз, фенилкетонурия) презентация
- Наследственные болезни и синдромы. Моногенные заболевания. Частые моногенные заболевания (муковисцидоз, фенилкетонурия)

Содержание
- 2. Входящий тестовый контроль по теме семинара 1. Наследственные болезни развиваются из-за: А) неблагоприятных внешних условий Б)
- 3. Рекомендуемые книги по теме семинара Роберт Л. Ньюссбаум, Родерик Р. Мак-Иннес, Хантингтон Ф. Виллард. Медицинская генетика.
- 4. Наследственные и мультифакториальные болезни Наследственные болезни – болезни, этиологическим фактором которых являются мутации. Мультифакториальные болезни –
- 5. Классификация наследственных болезней Хромосомные болезни – обусловлены геномными (изменение числа хромосом) или хромосомными (изменения структуры хромосом)
- 6. Особенности клинических проявлений наследственной патологии Семейный характер заболевания – есть сведения о похожем заболевании у родственников
- 7. Генотип и фенотип ГЕНОТИП – это сумма всех генов организма. Однако нередко понятие «генотип» используют для
- 8. Правило доминирования Из двух копий каждого гена, называемых аллелями и содержащихся в каждой клетке организма, одна
- 9. Некоторые символы, принятые при составлении родословной
- 10. Аутосомно-доминантный тип наследования
- 11. Аутосомно-доминантный тип наследования у каждого больного один из родителей обязательно болен (исключения: болезнь вызвана новой мутацией;);
- 12. Аутосомно-доминантный тип наследования Во-первых, один из родителей больных детей также должен быть болен. Во-вторых, болезнь должна
- 13. Аутосомно-рецессивный тип наследования
- 14. Носитель аутосомно-рецессивного признака является гомозиготой по мутантному аллелю гена (если мутантные аллели являются разными, то его
- 15. Если ген на Х-хромосоме отвечает за рецессивное заболевание, то сама мать должна быть здорова, так как
- 16. Появление и наследование мутаций Мозаицизм это существование в пределах одного организма генетически различающихся клеток. Является следствием
- 18. Цели и задачи медико-генетического консультирования Уточнение диагноза наследственного заболевания Определение типа наследования заболевания в семье Прогноз
- 19. Скрининг – комплекс мероприятий здравоохранения, начиная с выявления пациента до его вылечивания. Цель скрининга – повышение
- 20. Критерии неонатального скрининга Заболевание должно иметь четкие клинические и биохимические критерии Частота заболевания в данной популяции
- 21. Передача генетической информации в клетке экзоны интрон 1 интрон 2 К ДНК ДНК (экспрессирующийся ген) транскрипция
- 22. Под мутацией понимают все изменения в нуклеотидной последовательности ДНК, независимо от их локализации и влияния на
- 23. мутации нейтральные полиморфизмы патологические генные хромосомные точковые миссенс сайтов сплайсинга нонсенс регуляторные делеции и вставки со
- 24. КЛАССИФИКАЦИЯ МУТАЦИЙ. По месту локализации в различных типах ДНК-последовательности: мутации, затрагивающие кодирующие последовательности генов; мутации, затрагивающие
- 25. По типу изменения в нуклеотидной последовательности - мутации сдвига рамки считывания - возникают в результате вставки
- 26. По патогенетическому механизму - Мутации, ведущие к потере функции белка (loss-of-function) Мутации, ведущие к появлению новой
- 27. Классификация мутаций по клинической значимости BRCA database: 1. не патогенные или клинически не значимые; 2. вероятно
- 28. Номенклатура мутаций Обозначение аминокислот Примеры обозначения мутаций изменение в изменение в нуклеотидах аминокислотах миссенс А 263
- 29. Миссенс мутация - замена нуклеотида в ДНК, приводящая к заменен в белке одной аминокислоты на другую
- 30. Мутация сдвига рамки считывания Мутация сдвига рамки считывания — тип мутации в последовательности ДНК, для которого
- 32. Мутация сдвига рамки считывания Нормальная последовательность: ACC-AAC-TTC-ACC-TTG-… Thr Asn Phe Thr Leu … синтез нормального белка
- 34. Нейрофиброматоз I типа (болезнь Реклингхаузена) — это тяжелое системное наследственное заболевание с преимущественным поражением кожи и
- 35. Нейрофиброматоз тип 1 (болезнь Реклингаузена) Частота 1:3000-3500 нейрофибромы, глиома зрительного нерва, гемартромы радужной оболочки (узелки Лиша),
- 36. Нейрофиброматоз 1 типа Множественные нейрофибромы, гамартомы радужки, узелки Лиша - мелкие узелки размером с булавочную головку,
- 37. NF1 NF2 мерлин/шваномин нейрофибромин
- 39. Клиника нейрофиброматоза 1 типа
- 40. Диагностические критерии нейрофиброматоза 1 типа Диагноз может быть поставлен при наличии у больного по крайней мере
- 41. Диагностические критерии нейрофиброматоза типа II: • двусторонние невриномы VIII пары черепных нервов (по данным КТ или
- 42. NF1 NF2 мерлин/шваномин нейрофибромин
- 44. Генетическое тестирование Медико-генетическое консультирование больного и семьи
- 45. Сложности ДНК-диагностики факоматозов Несколько генов для одной болезни, например: нейрофиброматоз – NF1, NF2; туберозный склероз –
- 46. Высокая частота случаев мозаичных мутаций: NF1 – 10%, NF2 – 30%, TSC1/2 – 10%. Гетерогенность клинической
- 47. Мозаичная мутация при нейрофиброматозе Секвенирование по Сэнгеру. ДНК больного Контрольная ДНК Мутация или технический шум? Высокая
- 48. Синдром Ретта (RTT; OMIM 312750) генетически обусловленное прогрессирующее нейродегенеративное заболевание впервые описан в 1966 году A.
- 49. Клиника синдрома Ретта Диагностируется в возрасте 6 – 18 месяцев Нормальное пренатальное и перинатальное развитие Затем
- 50. Критерии диагностики синдрома Ретта Обязательные 1) нормальное пренатальное и перинатальное развитие 2) нормальное психомоторное развитие в
- 51. Белок MeCP2 – структура и функция Домены Метилсвязывающий, MBD Транскрипционно-репрессорный, TRD С-терминальный домен, C-term Ген картирован
- 52. 8 мажорных мутаций -70% всех случаев синдрома Ретта миссенс: R106W нонсенс: R168X R133W R255X T158M R270X
- 53. Мутации в гене MeCP2, описанные в литературе у мальчиков с умственной отсталостью Мутации в гене MeCP2
- 54. Инактивация Х-хромосомы (XCI) Неслучайная XCI встречается в норме в 1,5 - 3,5% при некоторых патологических состояниях:
- 55. Синдром есть, мутаций нет – причины? Мозаицизм Эпигенетические нарушения Генетическая гетерогенность: CDKL5/STK9 Тканеспецифическая экспрессия двух изоформ
- 56. Фенилкетонурия (ФКУ, PKU) фенилпировиноградная олигофрения MIM #261600 аутосомно-рецессивное заболевание частота заболевания 1 : 10000 в мире,
- 57. Патогенез ФКУ Значительное накопление в тканях и жидкостях больного фенилаланина и его производных (фенилпировиноградная, фенилмолочная, фенилуксусная,
- 58. Этиология ФКУ Классическая форма Атипичная форма (около 2%) Мутации в гене PAH Снижение или полное отсутствие
- 59. Ген PAH расположен на длинном плече 12 хромосомы в области q22-24.1 протяженностью около 90000 пар оснований
- 60. экзон 3 экзон 2 экзон 5 экзон 9 экзон 10 экзон 11 экзон 12 экзон 7
- 61. Диетотерапия
- 62. Частота встречаемости наиболее распространенных мутаций в России
- 64. Скачать презентацию

Входящий тестовый контроль по теме семинара
1. Наследственные болезни развиваются из-за:
А) неблагоприятных
Входящий тестовый контроль по теме семинара
1. Наследственные болезни развиваются из-за:
А) неблагоприятных

Рекомендуемые книги по теме семинара
Роберт Л. Ньюссбаум, Родерик Р. Мак-Иннес, Хантингтон
Рекомендуемые книги по теме семинара
Роберт Л. Ньюссбаум, Родерик Р. Мак-Иннес, Хантингтон
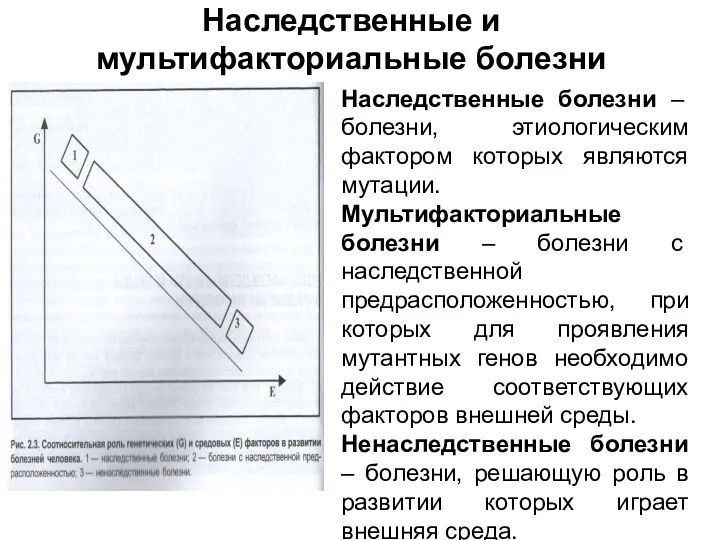
Наследственные и мультифакториальные болезни
Наследственные болезни – болезни, этиологическим фактором которых являются
Наследственные и мультифакториальные болезни
Наследственные болезни – болезни, этиологическим фактором которых являются

Классификация наследственных болезней
Хромосомные болезни – обусловлены геномными (изменение числа хромосом) или
Классификация наследственных болезней
Хромосомные болезни – обусловлены геномными (изменение числа хромосом) или

Особенности клинических проявлений наследственной патологии
Семейный характер заболевания – есть сведения о
Особенности клинических проявлений наследственной патологии
Семейный характер заболевания – есть сведения о

Генотип и фенотип
ГЕНОТИП – это сумма всех генов организма.
Однако нередко понятие
Генотип и фенотип
ГЕНОТИП – это сумма всех генов организма.
Однако нередко понятие

Правило доминирования
Из двух копий каждого гена, называемых аллелями и содержащихся в
Правило доминирования
Из двух копий каждого гена, называемых аллелями и содержащихся в
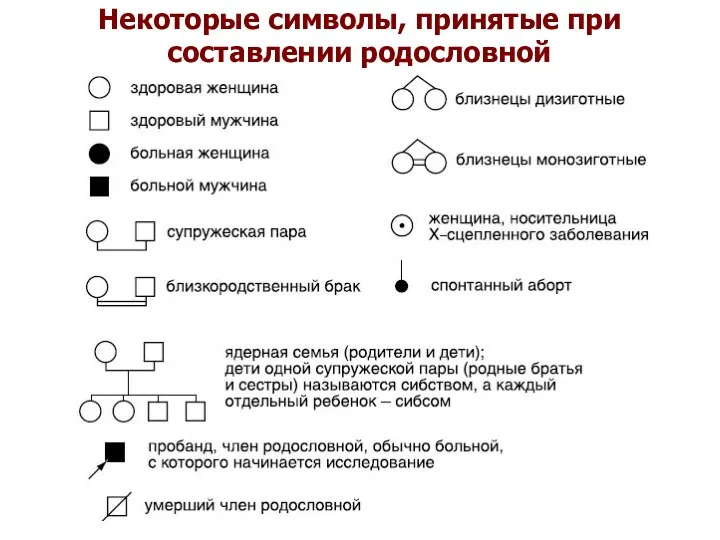
Некоторые символы, принятые при составлении родословной
Некоторые символы, принятые при составлении родословной
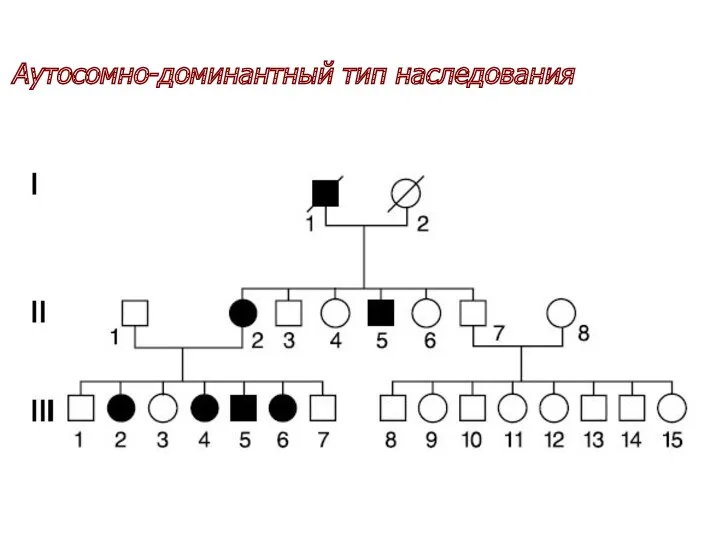
Аутосомно-доминантный тип наследования
Аутосомно-доминантный тип наследования
Аутосомно-доминантный тип наследования
у каждого больного один из родителей обязательно болен
Аутосомно-доминантный тип наследования
у каждого больного один из родителей обязательно болен

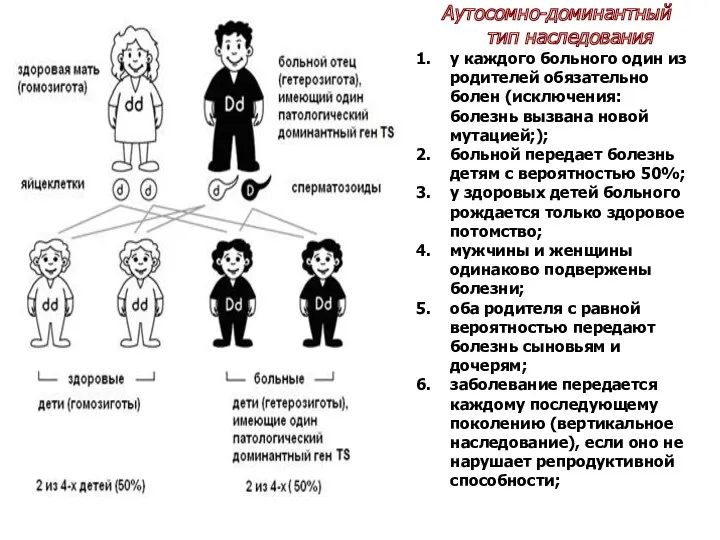
Аутосомно-доминантный тип наследования
Во-первых, один из родителей больных детей также должен быть
Аутосомно-доминантный тип наследования
Во-первых, один из родителей больных детей также должен быть
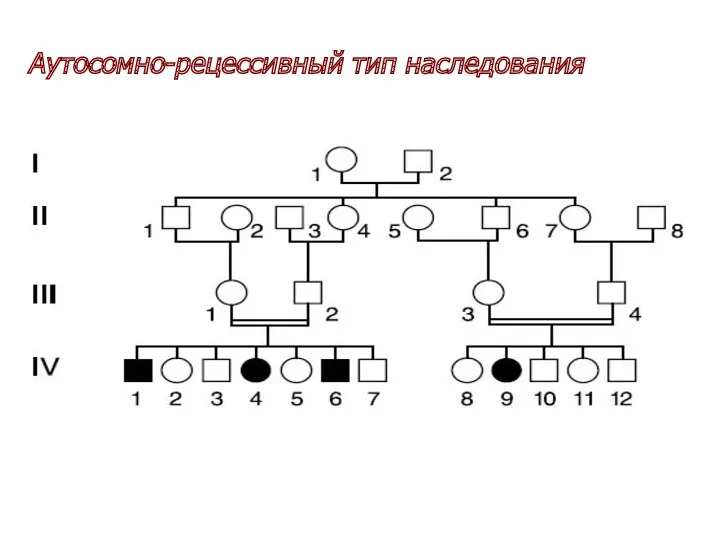
Аутосомно-рецессивный тип наследования
Аутосомно-рецессивный тип наследования

Носитель аутосомно-рецессивного признака является гомозиготой по мутантному аллелю гена (если мутантные
Носитель аутосомно-рецессивного признака является гомозиготой по мутантному аллелю гена (если мутантные
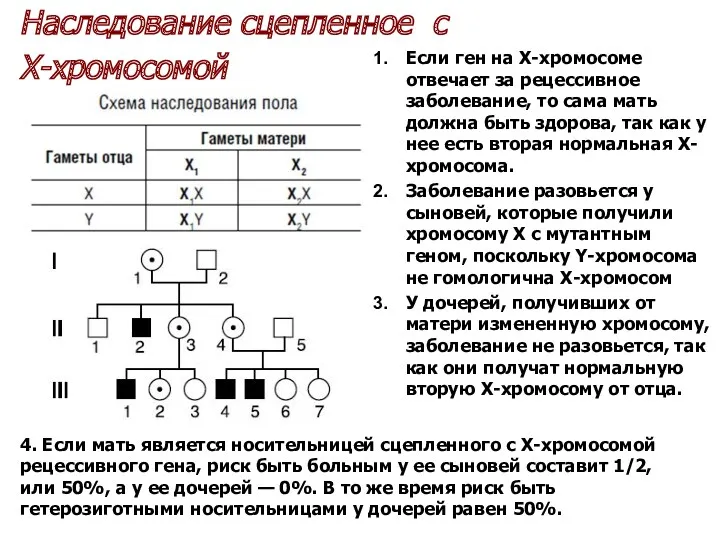
Если ген на Х-хромосоме отвечает за рецессивное заболевание, то сама мать
Если ген на Х-хромосоме отвечает за рецессивное заболевание, то сама мать
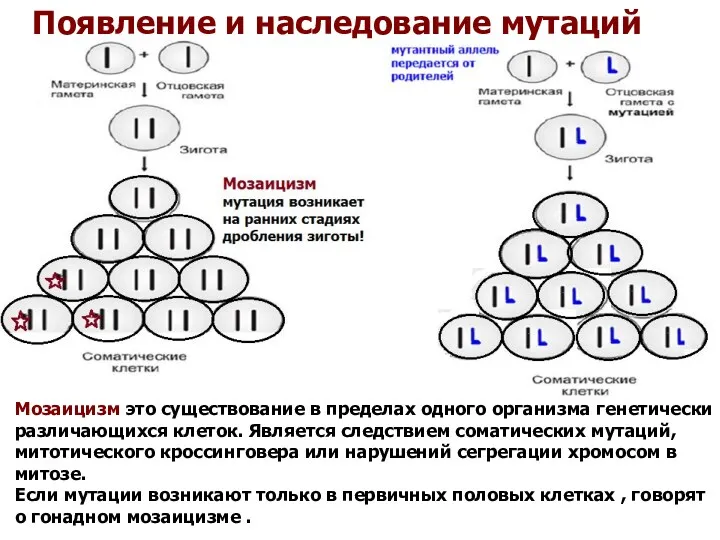
Появление и наследование мутаций
Мозаицизм это существование в пределах одного организма
Появление и наследование мутаций
Мозаицизм это существование в пределах одного организма


Цели и задачи медико-генетического консультирования
Уточнение диагноза наследственного заболевания
Определение типа наследования
Цели и задачи медико-генетического консультирования
Уточнение диагноза наследственного заболевания
Определение типа наследования

Скрининг – комплекс мероприятий здравоохранения, начиная с выявления пациента до его
Скрининг – комплекс мероприятий здравоохранения, начиная с выявления пациента до его

Критерии неонатального скрининга
Заболевание должно иметь четкие клинические и биохимические критерии
Частота
Критерии неонатального скрининга
Заболевание должно иметь четкие клинические и биохимические критерии
Частота
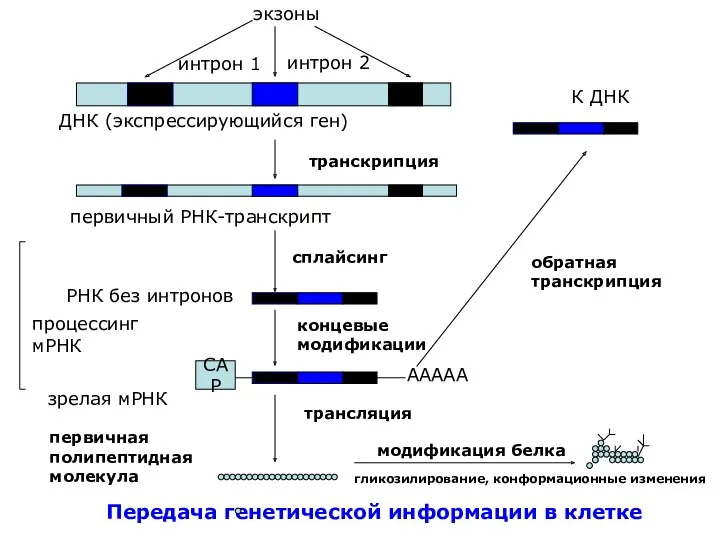
Передача генетической информации в клетке
экзоны
интрон 1
интрон 2
К ДНК
ДНК (экспрессирующийся ген)
транскрипция
первичный РНК-транскрипт
процессинг
мРНК
РНК
Передача генетической информации в клетке
экзоны
интрон 1
интрон 2
К ДНК
ДНК (экспрессирующийся ген)
транскрипция
первичный РНК-транскрипт
процессинг
мРНК
РНК

Под мутацией понимают все изменения в нуклеотидной последовательности ДНК, независимо от
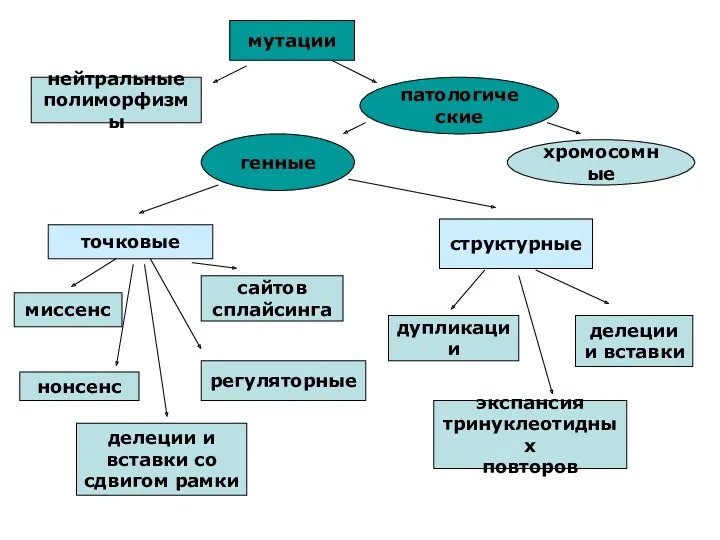
мутации
нейтральные
полиморфизмы
патологические
генные
хромосомные
точковые
миссенс
сайтов
сплайсинга
нонсенс
регуляторные
делеции и
вставки со
сдвигом рамки
структурные
дупликации
делеции
и вставки
экспансия
тринуклеотидных
повторов
мутации
нейтральные
полиморфизмы
патологические
генные
хромосомные
точковые
миссенс
сайтов
сплайсинга
нонсенс
регуляторные
делеции и
вставки со
сдвигом рамки
структурные
дупликации
делеции
и вставки
экспансия
тринуклеотидных
повторов

КЛАССИФИКАЦИЯ МУТАЦИЙ.
По месту локализации в различных типах ДНК-последовательности:
мутации, затрагивающие кодирующие последовательности
КЛАССИФИКАЦИЯ МУТАЦИЙ.
По месту локализации в различных типах ДНК-последовательности:
мутации, затрагивающие кодирующие последовательности

По типу изменения в нуклеотидной последовательности
- мутации сдвига рамки
- мутации сдвига рамки

По патогенетическому механизму
- Мутации, ведущие к потере функции белка (loss-of-function)
Мутации, ведущие
По патогенетическому механизму
- Мутации, ведущие к потере функции белка (loss-of-function)
Мутации, ведущие

Классификация мутаций по клинической значимости
BRCA database:
1. не патогенные или
Классификация мутаций по клинической значимости
BRCA database:
1. не патогенные или
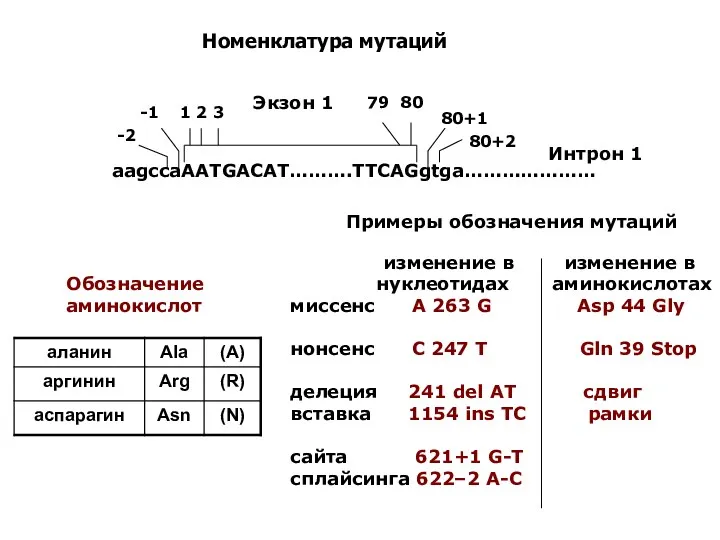
Номенклатура мутаций
Обозначение аминокислот
Примеры обозначения мутаций
изменение в изменение в
нуклеотидах
Номенклатура мутаций
Обозначение аминокислот
Примеры обозначения мутаций
изменение в изменение в
нуклеотидах
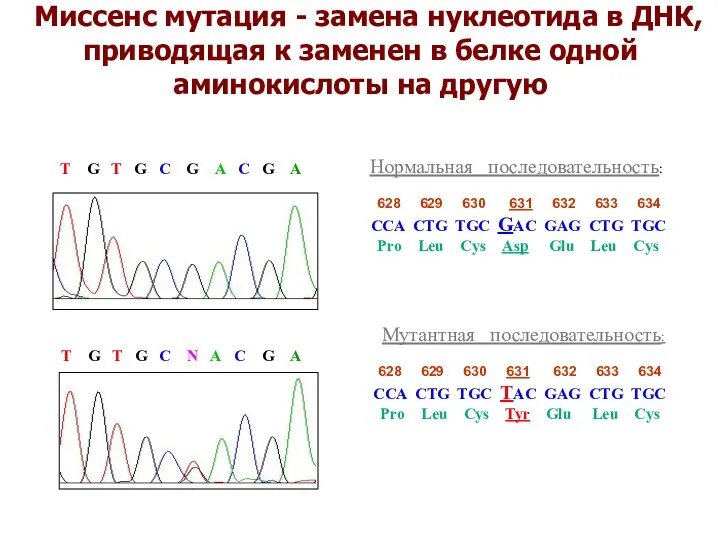
Миссенс мутация - замена нуклеотида в ДНК, приводящая к заменен
Миссенс мутация - замена нуклеотида в ДНК, приводящая к заменен
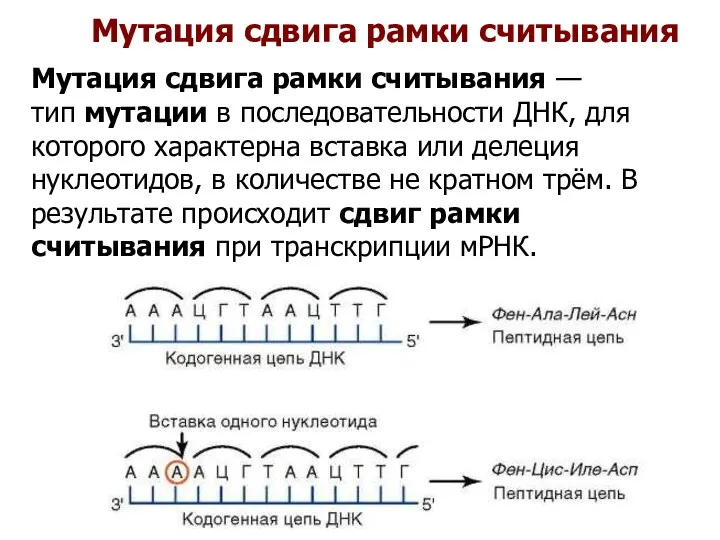
Мутация сдвига рамки считывания
Мутация сдвига рамки считывания — тип мутации в последовательности ДНК,
Мутация сдвига рамки считывания
Мутация сдвига рамки считывания — тип мутации в последовательности ДНК,
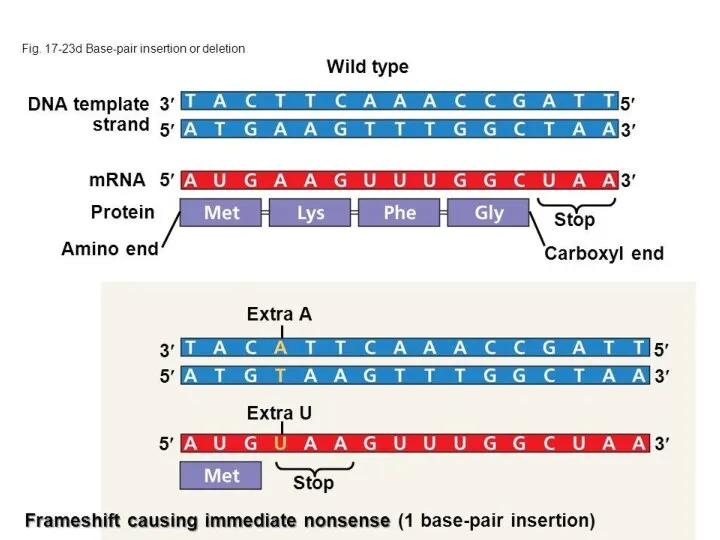
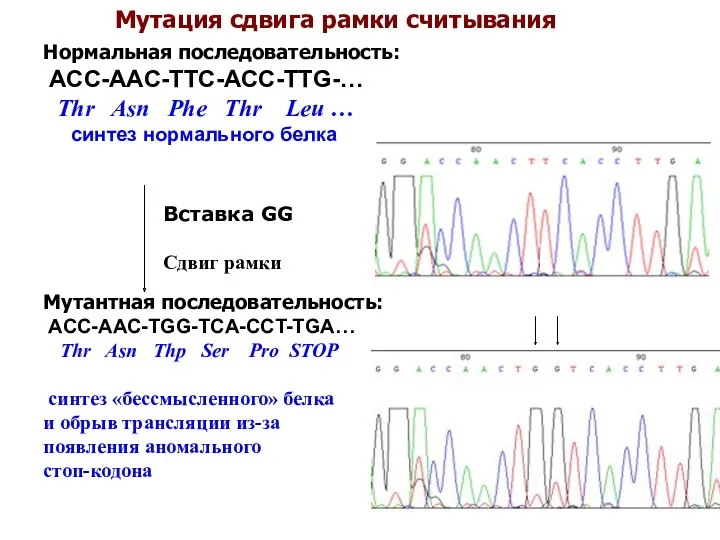
Мутация сдвига рамки считывания
Нормальная последовательность:
ACC-AAC-TTC-ACC-TTG-…
Thr Asn Phe Thr
Мутация сдвига рамки считывания
Нормальная последовательность:
ACC-AAC-TTC-ACC-TTG-…
Thr Asn Phe Thr


Нейрофиброматоз I типа (болезнь Реклингхаузена) —
это тяжелое системное наследственное заболевание
Нейрофиброматоз I типа (болезнь Реклингхаузена) —
это тяжелое системное наследственное заболевание
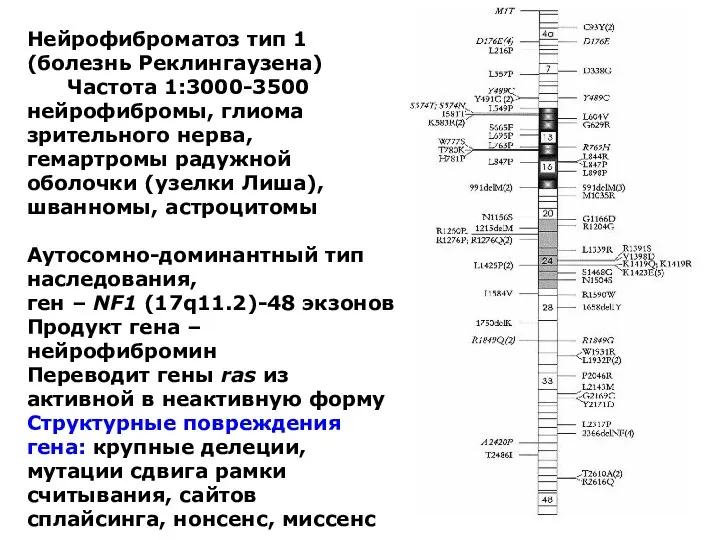
Нейрофиброматоз тип 1 (болезнь Реклингаузена)
Частота 1:3000-3500
нейрофибромы, глиома зрительного нерва, гемартромы
Нейрофиброматоз тип 1 (болезнь Реклингаузена)
Частота 1:3000-3500
нейрофибромы, глиома зрительного нерва, гемартромы
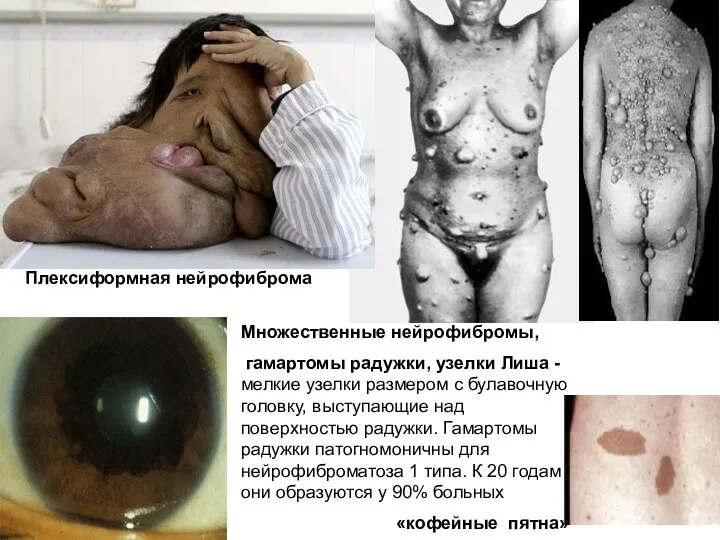
Нейрофиброматоз 1 типа
Множественные нейрофибромы,
гамартомы радужки, узелки Лиша - мелкие узелки
Нейрофиброматоз 1 типа
Множественные нейрофибромы,
гамартомы радужки, узелки Лиша - мелкие узелки
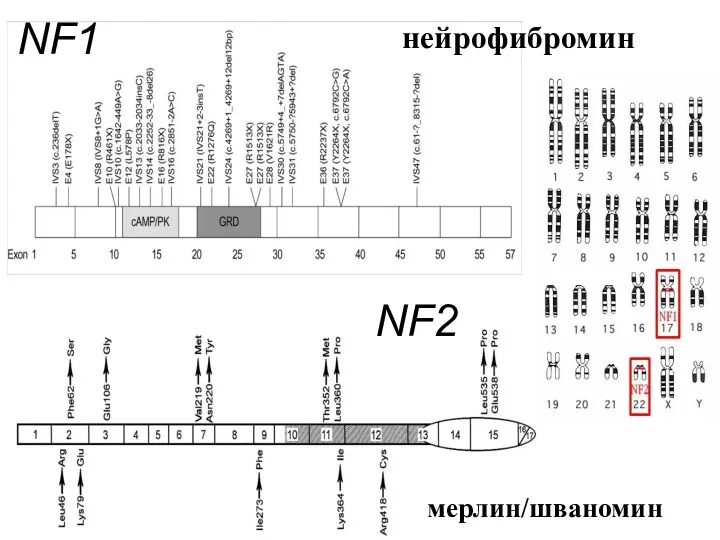
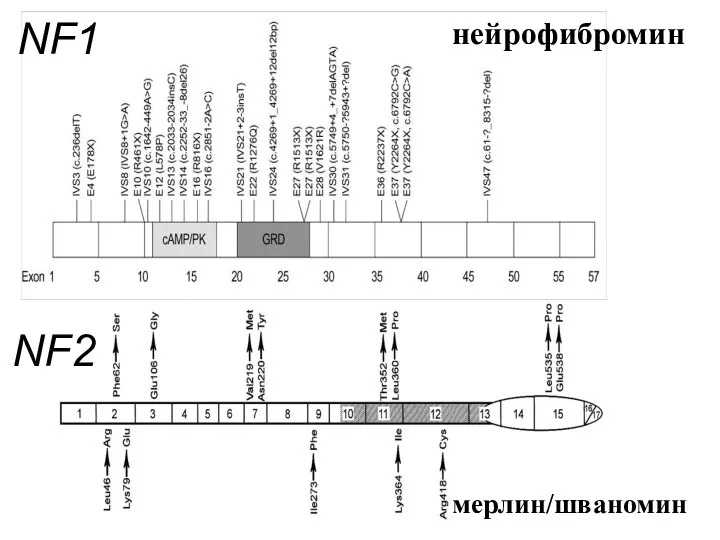
NF1
NF2
мерлин/шваномин
нейрофибромин
NF1
NF2
мерлин/шваномин
нейрофибромин
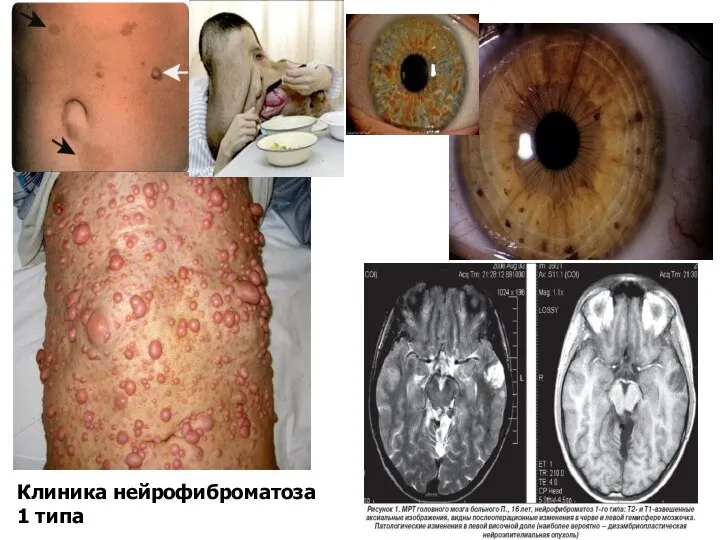
Клиника нейрофиброматоза 1 типа
Клиника нейрофиброматоза 1 типа

Диагностические критерии нейрофиброматоза 1 типа
Диагноз может быть поставлен при наличии у
Диагностические критерии нейрофиброматоза 1 типа
Диагноз может быть поставлен при наличии у

Диагностические критерии нейрофиброматоза
типа II:
• двусторонние невриномы VIII пары черепных
Диагностические критерии нейрофиброматоза
типа II:
• двусторонние невриномы VIII пары черепных
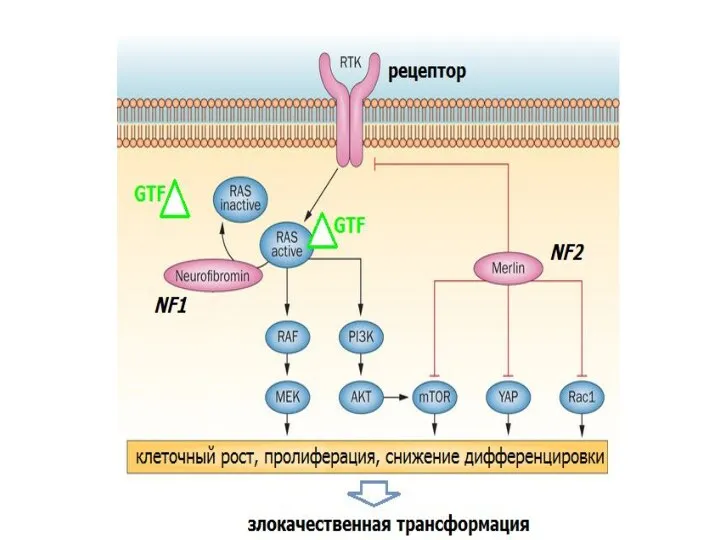
NF1
NF2
мерлин/шваномин
нейрофибромин
NF1
NF2
мерлин/шваномин
нейрофибромин

Генетическое тестирование
Медико-генетическое консультирование больного и семьи
Генетическое тестирование
Медико-генетическое консультирование больного и семьи

Сложности ДНК-диагностики факоматозов
Несколько генов для одной болезни, например:
нейрофиброматоз – NF1,
Сложности ДНК-диагностики факоматозов
Несколько генов для одной болезни, например:
нейрофиброматоз – NF1,

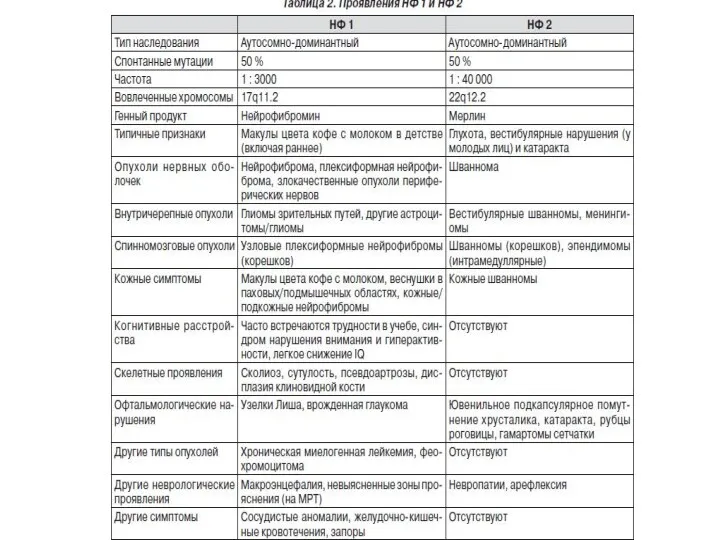
Высокая частота случаев мозаичных мутаций:
NF1 – 10%,
NF2 – 30%,
Высокая частота случаев мозаичных мутаций:
NF1 – 10%,
NF2 – 30%,
Мозаичная мутация при нейрофиброматозе
Секвенирование по Сэнгеру.
ДНК больного
Контрольная ДНК
Мутация или технический
Мозаичная мутация при нейрофиброматозе
Секвенирование по Сэнгеру.
ДНК больного
Контрольная ДНК
Мутация или технический

Синдром Ретта (RTT; OMIM 312750)
генетически обусловленное прогрессирующее нейродегенеративное заболевание
впервые
Синдром Ретта (RTT; OMIM 312750)
генетически обусловленное прогрессирующее нейродегенеративное заболевание
впервые

Клиника синдрома Ретта
Диагностируется в возрасте 6 – 18 месяцев
Нормальное пренатальное
Клиника синдрома Ретта
Диагностируется в возрасте 6 – 18 месяцев
Нормальное пренатальное

Критерии диагностики синдрома Ретта
Обязательные
1) нормальное пренатальное и перинатальное развитие
2) нормальное
Критерии диагностики синдрома Ретта
Обязательные
1) нормальное пренатальное и перинатальное развитие
2) нормальное
Белок MeCP2 – структура и функция
Домены
Метилсвязывающий, MBD
Транскрипционно-репрессорный, TRD
С-терминальный домен, C-term
Белок MeCP2 – структура и функция
Домены
Метилсвязывающий, MBD
Транскрипционно-репрессорный, TRD
С-терминальный домен, C-term
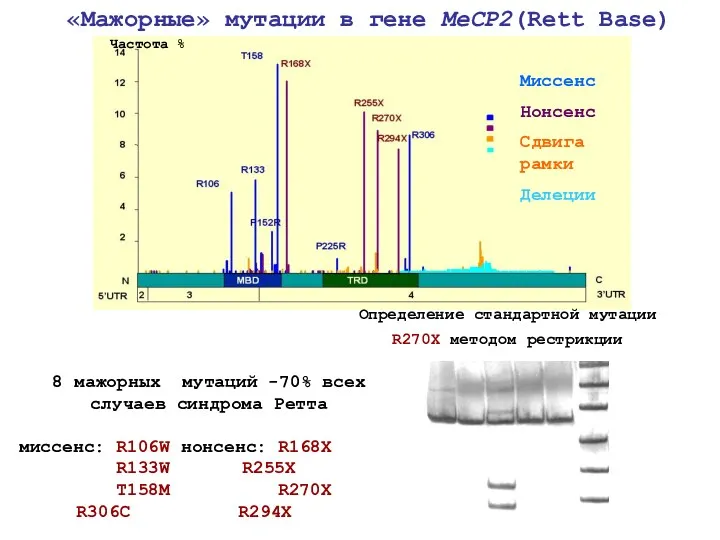
8 мажорных мутаций -70% всех случаев синдрома Ретта
миссенс: R106W нонсенс: R168X
8 мажорных мутаций -70% всех случаев синдрома Ретта
миссенс: R106W нонсенс: R168X
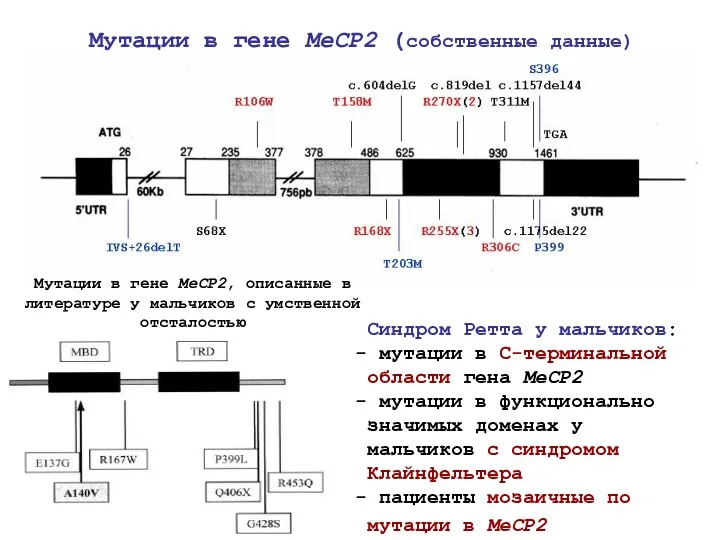
Мутации в гене MeCP2, описанные в литературе у мальчиков с умственной
Мутации в гене MeCP2, описанные в литературе у мальчиков с умственной

Инактивация Х-хромосомы (XCI)
Неслучайная XCI встречается
в норме в 1,5 -
Инактивация Х-хромосомы (XCI)
Неслучайная XCI встречается
в норме в 1,5 -

Синдром есть, мутаций нет – причины?
Мозаицизм
Эпигенетические нарушения
Генетическая гетерогенность: CDKL5/STK9
Тканеспецифическая
Синдром есть, мутаций нет – причины?
Мозаицизм
Эпигенетические нарушения
Генетическая гетерогенность: CDKL5/STK9
Тканеспецифическая

Фенилкетонурия (ФКУ, PKU)
фенилпировиноградная олигофрения
MIM #261600
аутосомно-рецессивное заболевание
частота заболевания 1 :
Фенилкетонурия (ФКУ, PKU)
фенилпировиноградная олигофрения
MIM #261600
аутосомно-рецессивное заболевание
частота заболевания 1 :
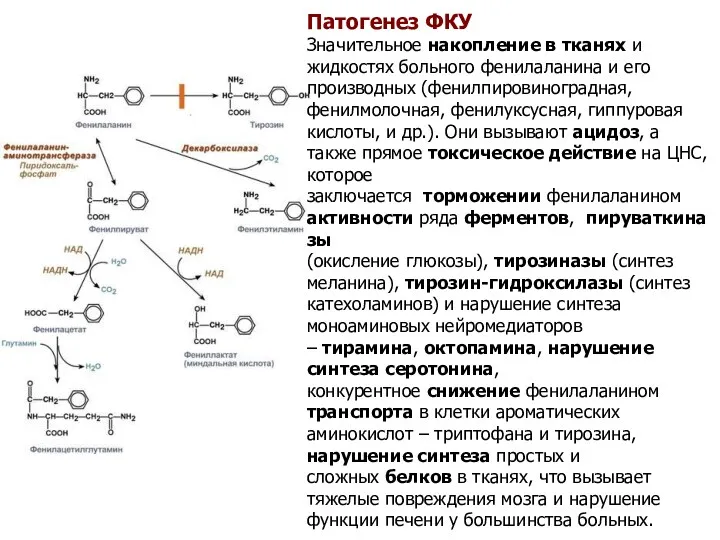
Патогенез ФКУ
Значительное накопление в тканях и жидкостях больного фенилаланина и его производных (фенилпировиноградная,
Патогенез ФКУ
Значительное накопление в тканях и жидкостях больного фенилаланина и его производных (фенилпировиноградная,

Этиология
ФКУ
Классическая форма
Атипичная форма (около 2%)
Мутации в гене PAH
Снижение или полное отсутствие
Этиология
ФКУ
Классическая форма
Атипичная форма (около 2%)
Мутации в гене PAH
Снижение или полное отсутствие
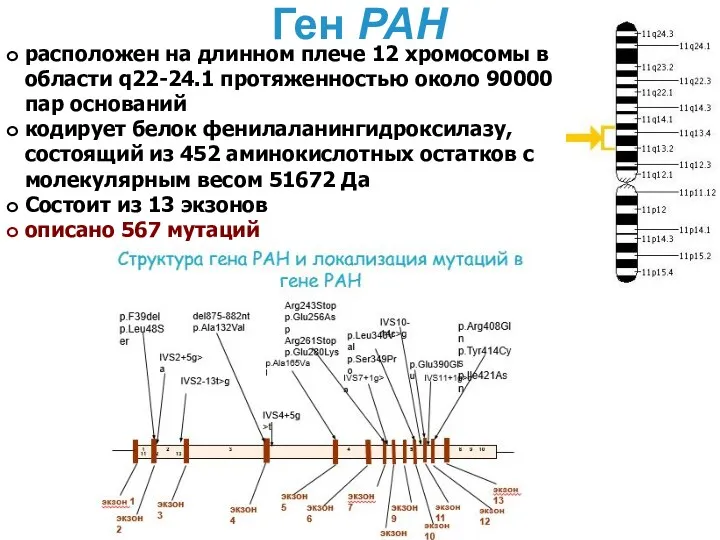
Ген PAH
расположен на длинном плече 12 хромосомы в области q22-24.1 протяженностью
Ген PAH
расположен на длинном плече 12 хромосомы в области q22-24.1 протяженностью
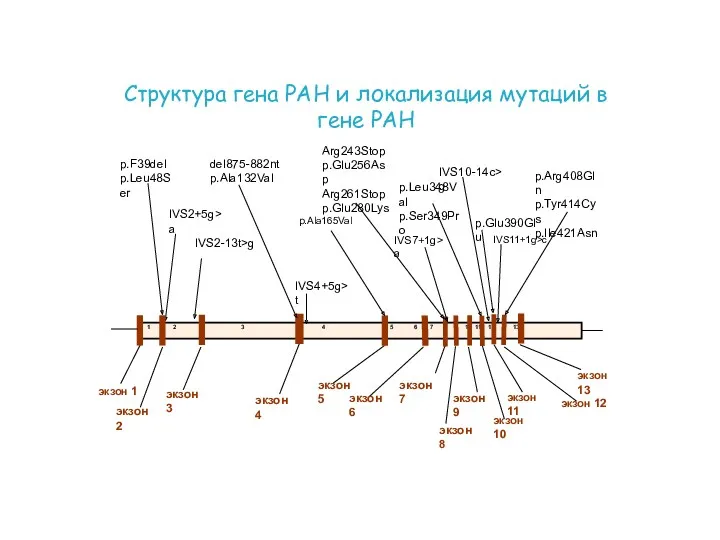
экзон 3
экзон 2
экзон 5
экзон 9
экзон 10
экзон 11
экзон 12
экзон 7
экзон 8
экзон 6
экзон
экзон 3
экзон 2
экзон 5
экзон 9
экзон 10
экзон 11
экзон 12
экзон 7
экзон 8
экзон 6
экзон

Диетотерапия
Диетотерапия
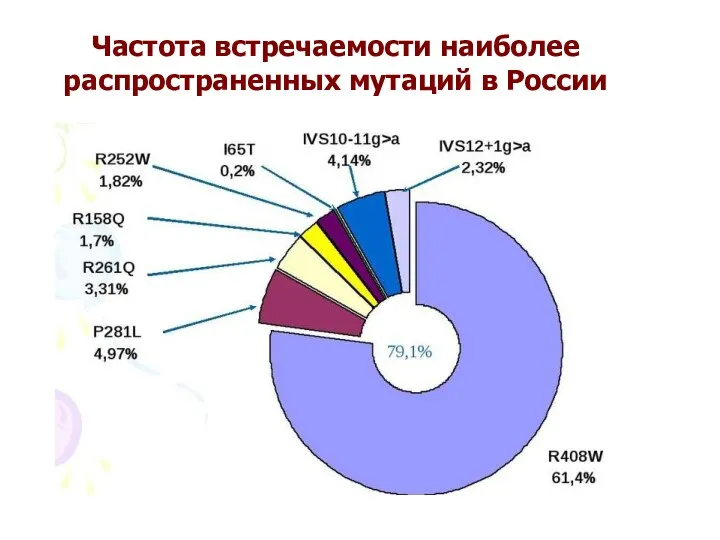
Частота встречаемости наиболее распространенных мутаций в России
Частота встречаемости наиболее распространенных мутаций в России
 Выращивание овощей в домашних условиях
Выращивание овощей в домашних условиях История возникновения и развития живого на Земле
История возникновения и развития живого на Земле Внешнее строение листа. Жилкование
Внешнее строение листа. Жилкование Занимательное путешествие в мир насекомых
Занимательное путешествие в мир насекомых Необычный сахар
Необычный сахар Удивительные овощи. Фото-отчет по выставке
Удивительные овощи. Фото-отчет по выставке Микрооргнаизмдерді дақылдауға арналған қоректік орталар, қоректік орталарды залалсыздандыру әдістері
Микрооргнаизмдерді дақылдауға арналған қоректік орталар, қоректік орталарды залалсыздандыру әдістері Ластоногие. Общая характеристика ластоногих
Ластоногие. Общая характеристика ластоногих Птахи. Загальна характеристика птахів
Птахи. Загальна характеристика птахів Обучающая презентация Флора г. Аксая Ростовской области
Обучающая презентация Флора г. Аксая Ростовской области Решение задач по генетике
Решение задач по генетике Птицы, обитающие на территории Русского острова
Птицы, обитающие на территории Русского острова Антропология
Антропология Внутрішнє вухо, його частини, топографія, кістковий лабіринт
Внутрішнє вухо, його частини, топографія, кістковий лабіринт Подготовка учащихся к государственной итоговой аттестации по биологии
Подготовка учащихся к государственной итоговой аттестации по биологии Органы цветковых растений. Корень
Органы цветковых растений. Корень Когда жили динозавры? Окружающий мир. 1 класс
Когда жили динозавры? Окружающий мир. 1 класс Опоно-рухова система
Опоно-рухова система Красная книга Казахстана
Красная книга Казахстана Прийшла весна - тепло принесла!
Прийшла весна - тепло принесла! Презентация Биография И. Н. Сеченова, 8 кл
Презентация Биография И. Н. Сеченова, 8 кл Всемирный день защиты животных
Всемирный день защиты животных Анимированный тест Введение в курс общей биологии 10 класс
Анимированный тест Введение в курс общей биологии 10 класс Общие пути катаболизма. Дыхательная цепь
Общие пути катаболизма. Дыхательная цепь Разработка интегрированного урока биологии и географии по теме История формирования сообществ живых организмов. Биогеография. Основные биомы суши для 11 класса
Разработка интегрированного урока биологии и географии по теме История формирования сообществ живых организмов. Биогеография. Основные биомы суши для 11 класса Парша яблони и меры борьбы с ней в условиях Уйгурского района Алматинской области
Парша яблони и меры борьбы с ней в условиях Уйгурского района Алматинской области Чарльз Дарвин и его теория эволюции
Чарльз Дарвин и его теория эволюции Комахи (природознавство, 3 клас)
Комахи (природознавство, 3 клас)