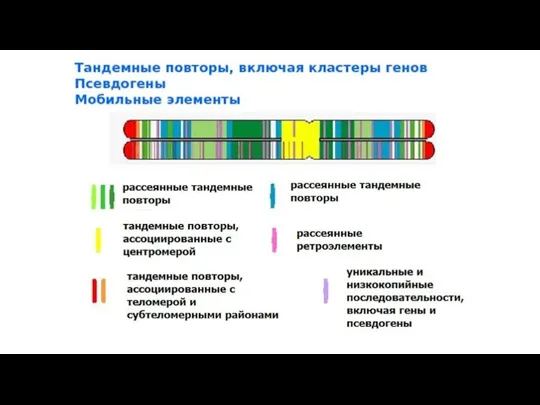
Повторяющиеся элементы в геноме человека. Механизмы экспансии тринуклеотидных повторов. Характеристика болезней экспансии презентация
- Повторяющиеся элементы в геноме человека. Механизмы экспансии тринуклеотидных повторов. Характеристика болезней экспансии

Содержание
- 3. Классическая работа Бриттена и Кона (Britten, Kohne, 1968) по кинетике ренатурации ДНК показала, что геномы высших
- 4. 5% - кодирующая ДНК 15% - сателлитная ДНК 10% - Alu-повторы 20% - LINE-повторы 15% -
- 5. Классификация повторов в кодирующей ДНК
- 6. Мультигенные семейства В классические семейства объединяют гены, отличающиеся высокой гомологией по всей длине или, по крайней
- 7. Кластеры генов Некоторые генные семейства расположены компактно на одной или нескольких хромосомах, самое крупное из которых
- 8. Рассеянные генные семейства Рассеянные семейства в зависимости от происхождения разделяют на: Семейства генов, произошедших из разных
- 9. Псевдогены Псевдогены – копии генов с утраченной функцией. Если они сохраняют экзон-интронную структуру исходного гена, то
- 10. Образование процессированного псевдогена
- 11. Клиническое значение образования псевдогенов Тандемное повторение гомологичных последовательностей генов и псевдогенов в пределах кластеров способствует повышению
- 12. Рассеянные некодирующие повторы Подавляющее большинство рассеянных некодирующих повторов – мобильные элементы генома – транспозоны и, в
- 13. Короткие рассеянные повторы SINE – short interspersed nuclear elements. Средняя длина коровой единицы 100-300 п.н. (например,
- 14. Эффекты инсерции Alu-повторов Инсерции Alu-элементов составляют 0,1% от общего числа мутаций, приводящих к заболеваниям человека.
- 16. Повторяющиеся последовательности в геноме VNTR (variable number tandem repeats)
- 19. Репликация повторяющихся последовательностей
- 20. Модель экспансии повторов на запаздывающей цепи ДНК
- 21. Представленность отдельных аминокислот в общей структуре гомополиаминокислотных трактов протеома человека
- 22. Мутантные повторы проявляют как мейотическую, так и митотическую нестабильность, c увеличением, а не сокращением числа повторяющихся
- 23. Болезни экспансии тринуклеотидных повторов Экспансия кодирующих тринуклеотидных повторов CAG-повторы – полиглутаминовые тракты Хорея Гентингтона GCG,GCA,GCT,GCC-повторы- полиаланиновые
- 25. Несмотря на полное отсутствие гомологии между белками, содержащими полиглутаминовые тракты, болезни экспансии кодирующих CAG-повторов обладают общими
- 26. Патогенез заболеваний экспансии полиглутаминовых трактов Белки, содержащие увеличенные полиглутаминовые тракты, преобретают новую цитотоксическую функцию (мутации типа
- 27. Болезни экспансии полиглутаминовых трактов CAG/CAA
- 28. Ген, кодирующий андрогеновый рецептор (AR/HUMARA), локализован на длинном плече хромосомы Х (локус Xq11-12) и содержит 8
- 29. Андрогеновый рецептор
- 30. Полиморфизм CAG-повтора гена AR ≤18 19-25 26-35 38-62 повышен риск возникновения рака простаты наиболее частые варианты
- 31. Спинально - бульбарная амиотрофия Кеннеди Названа по имени американского невролога W. Kennedy, описавшего её в 1968
- 32. 9-36 CAG норма 51 CAG носитель 38-62 CAG СБА
- 33. Фрагментный анализ CAG-повтора AR с помощью GeneMapper v. 3.5.
- 34. Болезни экспансии полиглутаминовых трактов CAG/CAA
- 35. Хорея Гентингтона
- 36. Хорея Гентингтона - нейродегенеративное наследственное заболевание. Хорея - форма гиперкинеза, характеризуется непроизвольными, быстрыми, нерегулируемыми движениями в
- 37. Причины болезни Гентингтона Ген болезни Гентингтона располагается в 4р16.3 , содержит 67 экзонов и кодирует белок
- 38. Хорея Гентингтона
- 39. Болезни экспансии полиглутаминовых трактов CAG/CAA
- 40. Спиноцеребеллярные атаксии В основе этой группы заболеваний лежат прогрессирующие дегенеративные изменения в нейронах мозжечка, головного мозга
- 41. Спиноцеребеллярная атаксия, типы 1 и 2 Оба заболевания начинаются в 30-40 лет с появления легкого нарушения
- 42. Диагностика При КТ мозга наблюдается истончение средней ножки мозжечка, расширение субарахноидального пространства полушарий и червя мозжечка,
- 43. Болезни экспансии полиаланиновых трактов GCG, GCA, GCT, GCC
- 45. Скачать презентацию
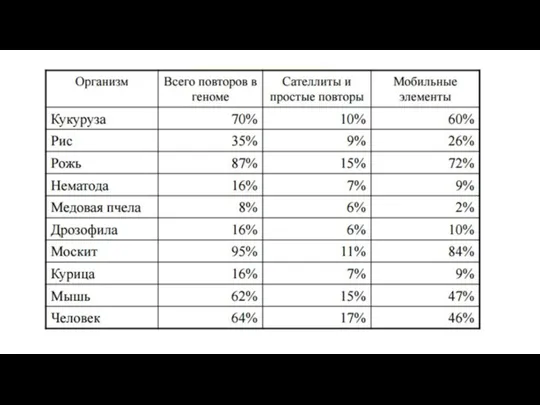

Классическая работа Бриттена и Кона (Britten, Kohne, 1968) по кинетике ренатурации ДНК показала,
Классическая работа Бриттена и Кона (Britten, Kohne, 1968) по кинетике ренатурации ДНК показала,
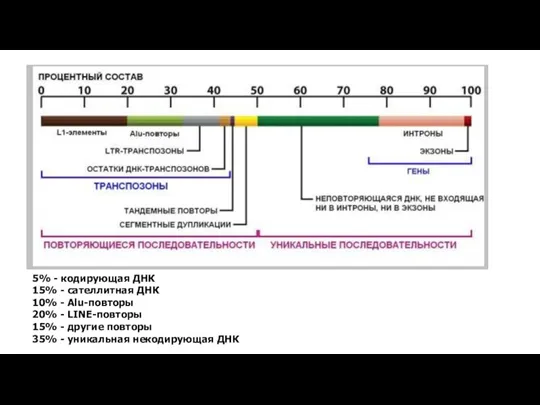
5% - кодирующая ДНК
15% - сателлитная ДНК
10% - Alu-повторы
20% - LINE-повторы
15%
5% - кодирующая ДНК
15% - сателлитная ДНК
10% - Alu-повторы
20% - LINE-повторы
15%
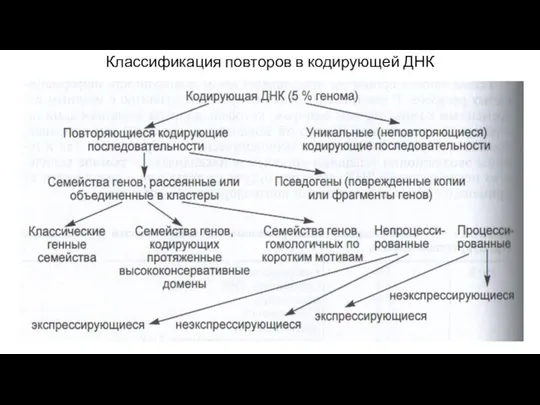
Классификация повторов в кодирующей ДНК
Классификация повторов в кодирующей ДНК

Мультигенные семейства
В классические семейства объединяют гены, отличающиеся высокой гомологией по всей
Мультигенные семейства
В классические семейства объединяют гены, отличающиеся высокой гомологией по всей
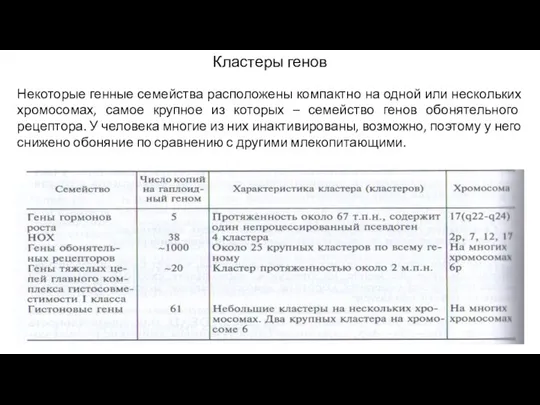
Кластеры генов
Некоторые генные семейства расположены компактно на одной или нескольких хромосомах,
Кластеры генов
Некоторые генные семейства расположены компактно на одной или нескольких хромосомах,
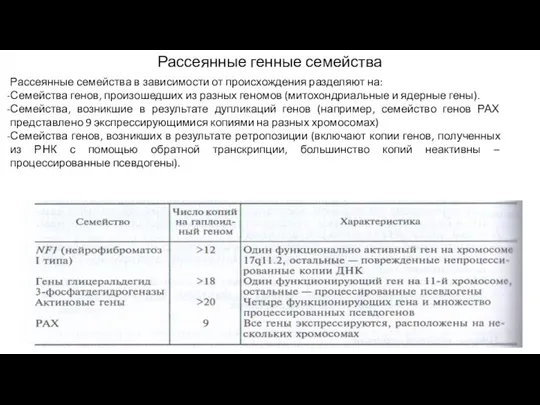
Рассеянные генные семейства
Рассеянные семейства в зависимости от происхождения разделяют на:
Семейства генов,
Рассеянные генные семейства
Рассеянные семейства в зависимости от происхождения разделяют на:
Семейства генов,

Псевдогены
Псевдогены – копии генов с утраченной функцией. Если они сохраняют экзон-интронную
Псевдогены
Псевдогены – копии генов с утраченной функцией. Если они сохраняют экзон-интронную
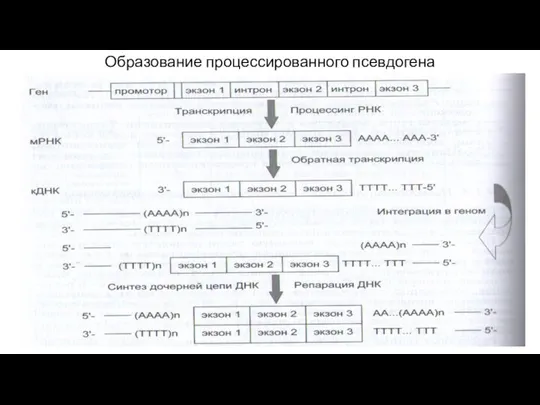
Образование процессированного псевдогена
Образование процессированного псевдогена

Клиническое значение образования псевдогенов
Тандемное повторение гомологичных последовательностей генов и псевдогенов в
Клиническое значение образования псевдогенов
Тандемное повторение гомологичных последовательностей генов и псевдогенов в

Рассеянные некодирующие повторы
Подавляющее большинство рассеянных некодирующих повторов – мобильные элементы генома
Рассеянные некодирующие повторы
Подавляющее большинство рассеянных некодирующих повторов – мобильные элементы генома

Короткие рассеянные повторы
SINE – short interspersed nuclear elements. Средняя длина коровой
Короткие рассеянные повторы
SINE – short interspersed nuclear elements. Средняя длина коровой

Эффекты инсерции Alu-повторов
Инсерции Alu-элементов составляют 0,1% от общего числа мутаций, приводящих
Эффекты инсерции Alu-повторов
Инсерции Alu-элементов составляют 0,1% от общего числа мутаций, приводящих
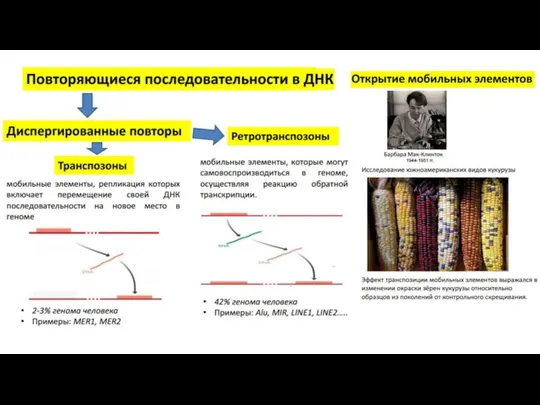
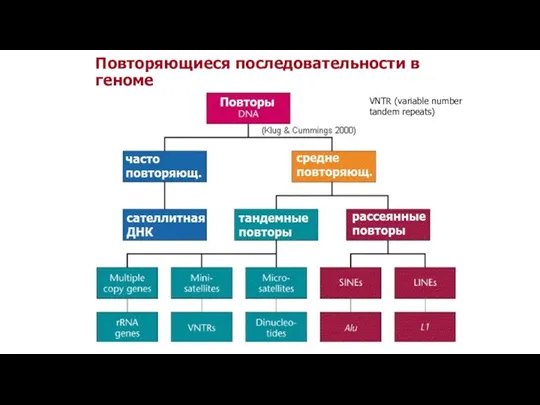
Повторяющиеся последовательности в геноме
VNTR (variable number tandem repeats)
Повторяющиеся последовательности в геноме
VNTR (variable number tandem repeats)

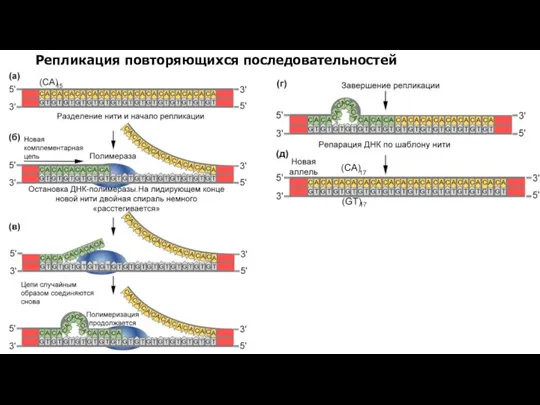
Репликация повторяющихся последовательностей
Репликация повторяющихся последовательностей

Модель экспансии повторов
на запаздывающей цепи ДНК
Модель экспансии повторов
на запаздывающей цепи ДНК

Представленность отдельных аминокислот в общей структуре гомополиаминокислотных трактов протеома человека
Представленность отдельных аминокислот в общей структуре гомополиаминокислотных трактов протеома человека

Мутантные повторы проявляют как мейотическую, так и митотическую нестабильность, c
Мутантные повторы проявляют как мейотическую, так и митотическую нестабильность, c

Болезни экспансии тринуклеотидных повторов
Экспансия кодирующих тринуклеотидных повторов
CAG-повторы – полиглутаминовые тракты
Болезни экспансии тринуклеотидных повторов
Экспансия кодирующих тринуклеотидных повторов
CAG-повторы – полиглутаминовые тракты
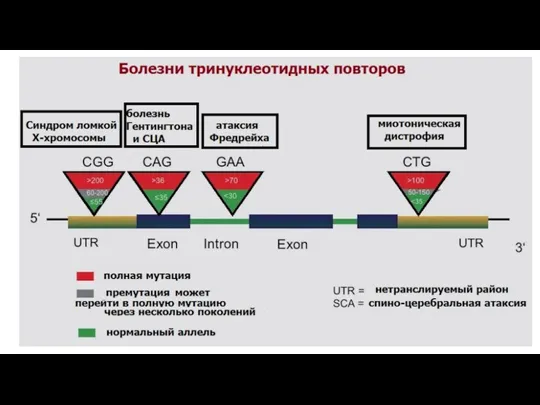

Несмотря на полное отсутствие гомологии между белками, содержащими полиглутаминовые тракты, болезни
Несмотря на полное отсутствие гомологии между белками, содержащими полиглутаминовые тракты, болезни

Патогенез заболеваний экспансии полиглутаминовых трактов
Белки, содержащие увеличенные полиглутаминовые тракты, преобретают новую
Патогенез заболеваний экспансии полиглутаминовых трактов
Белки, содержащие увеличенные полиглутаминовые тракты, преобретают новую
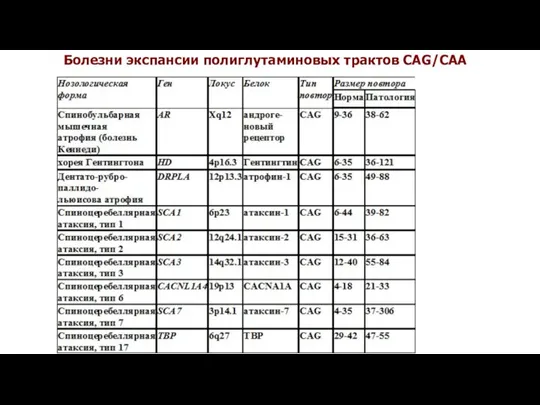
Болезни экспансии полиглутаминовых трактов CAG/CAA
Болезни экспансии полиглутаминовых трактов CAG/CAA

Ген, кодирующий андрогеновый рецептор (AR/HUMARA), локализован на длинном плече хромосомы Х
Ген, кодирующий андрогеновый рецептор (AR/HUMARA), локализован на длинном плече хромосомы Х
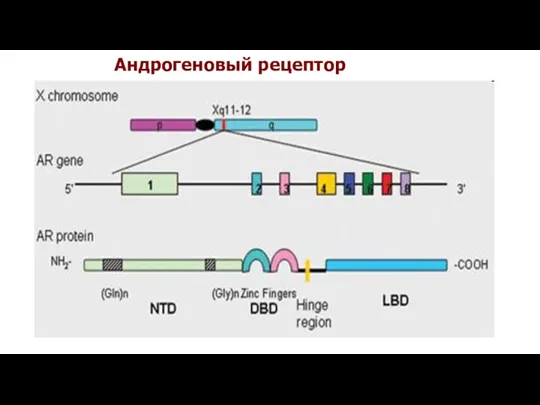
Андрогеновый рецептор
Андрогеновый рецептор

Полиморфизм CAG-повтора гена AR
≤18
19-25
26-35
38-62
повышен риск
возникновения
рака простаты
наиболее частые
варианты нормы
повышен риск
возникновения
олиго- и
азооспермии
SBMA
Полиморфизм CAG-повтора гена AR
≤18
19-25
26-35
38-62
повышен риск
возникновения
рака простаты
наиболее частые
варианты нормы
повышен риск
возникновения
олиго- и
азооспермии
SBMA

Спинально - бульбарная амиотрофия Кеннеди
Названа по имени американского невролога W. Kennedy,
Спинально - бульбарная амиотрофия Кеннеди
Названа по имени американского невролога W. Kennedy,
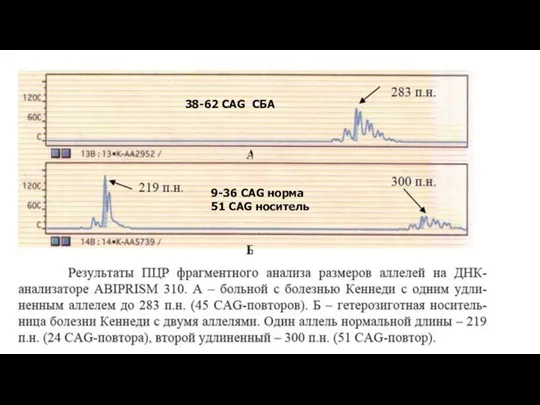
9-36 CAG норма
51 CAG носитель
38-62 CAG СБА
9-36 CAG норма
51 CAG носитель
38-62 CAG СБА
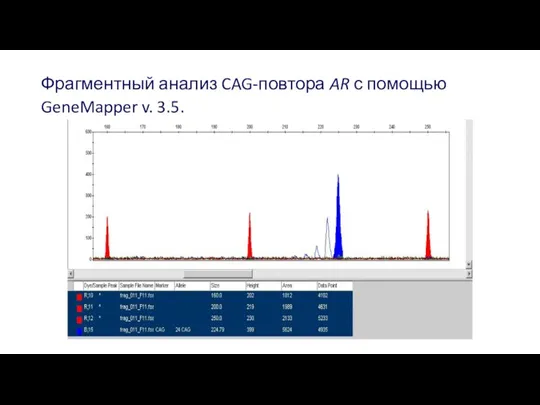
Фрагментный анализ CAG-повтора AR с помощью GeneMapper v. 3.5.
Фрагментный анализ CAG-повтора AR с помощью GeneMapper v. 3.5.
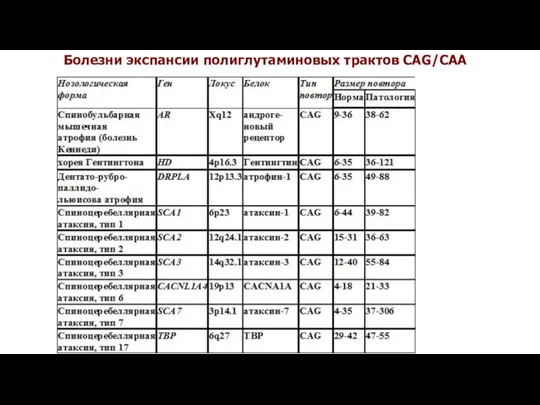
Болезни экспансии полиглутаминовых трактов CAG/CAA
Болезни экспансии полиглутаминовых трактов CAG/CAA
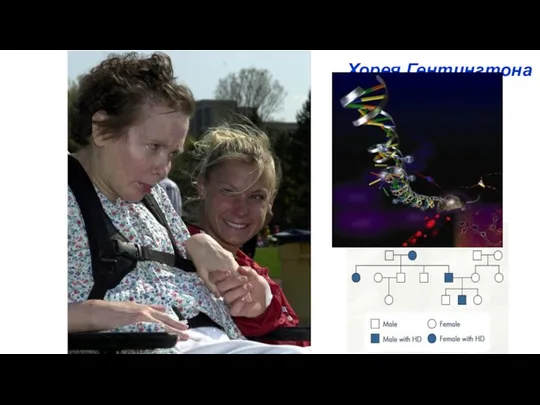
Хорея Гентингтона
Хорея Гентингтона

Хорея Гентингтона - нейродегенеративное наследственное заболевание.
Хорея - форма гиперкинеза, характеризуется
Хорея Гентингтона - нейродегенеративное наследственное заболевание.
Хорея - форма гиперкинеза, характеризуется

Причины болезни Гентингтона
Ген болезни Гентингтона располагается в 4р16.3 , содержит
Причины болезни Гентингтона
Ген болезни Гентингтона располагается в 4р16.3 , содержит
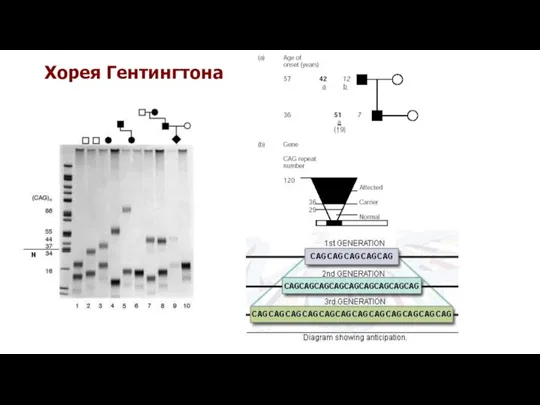
Хорея Гентингтона
Хорея Гентингтона
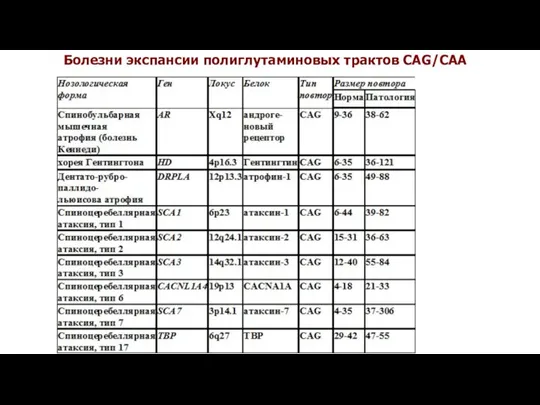
Болезни экспансии полиглутаминовых трактов CAG/CAA
Болезни экспансии полиглутаминовых трактов CAG/CAA

Спиноцеребеллярные атаксии
В основе этой группы заболеваний лежат прогрессирующие дегенеративные изменения в
Спиноцеребеллярные атаксии
В основе этой группы заболеваний лежат прогрессирующие дегенеративные изменения в

Спиноцеребеллярная атаксия, типы 1 и 2
Оба заболевания начинаются в 30-40 лет
Спиноцеребеллярная атаксия, типы 1 и 2
Оба заболевания начинаются в 30-40 лет

Диагностика
При КТ мозга наблюдается истончение
средней ножки мозжечка, расширение субарахноидального пространства
Диагностика
При КТ мозга наблюдается истончение
средней ножки мозжечка, расширение субарахноидального пространства
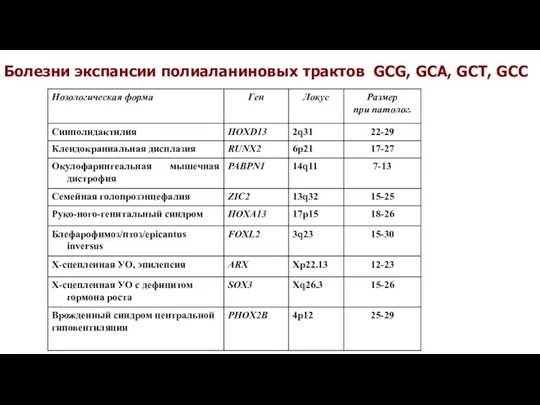
Болезни экспансии полиаланиновых трактов GCG, GCA, GCT, GCC
Болезни экспансии полиаланиновых трактов GCG, GCA, GCT, GCC
 Значення та охорона членистоногих
Значення та охорона членистоногих Особенности строения прокариотической клетки
Особенности строения прокариотической клетки Симбиотические организмы лишайники
Симбиотические организмы лишайники Вода, її роль у життєдіяльності організмів
Вода, її роль у життєдіяльності організмів Прокариотты және эукариотты өсімдіктер клеткасы
Прокариотты және эукариотты өсімдіктер клеткасы Мы в ответе за тех, кого приручили
Мы в ответе за тех, кого приручили Внешнее строение листа
Внешнее строение листа Строение, виды и значение вирусов. Прионы. Вироиды
Строение, виды и значение вирусов. Прионы. Вироиды Покормите птиц 2020. Эколого-культурная акция
Покормите птиц 2020. Эколого-культурная акция Гигиена органов пищеварения. Предупреждение желудочно–кишечных инфекций
Гигиена органов пищеварения. Предупреждение желудочно–кишечных инфекций Ткани животных
Ткани животных Простые и сложные соцветия
Простые и сложные соцветия Дәріс №2. Жасушалық технологияның негізі– жасуша өсіндісі
Дәріс №2. Жасушалық технологияның негізі– жасуша өсіндісі Введение генов в клетки растений - основные способы
Введение генов в клетки растений - основные способы Воздействие человека и его деятельности на животных
Воздействие человека и его деятельности на животных Строение, размножение и развитие рыб
Строение, размножение и развитие рыб Функциональная анатомия мышц конечностей
Функциональная анатомия мышц конечностей Физиология пищеварения
Физиология пищеварения Семейство зайцев. Школьная научно практическая конференция
Семейство зайцев. Школьная научно практическая конференция Развитие и устойчивость экосистем
Развитие и устойчивость экосистем Регуляция кровообращения
Регуляция кровообращения Способи годівлі птахів
Способи годівлі птахів Методы биологических исследований
Методы биологических исследований Промежуточные филаменты
Промежуточные филаменты Ақтөбеде сирек кездесетін өсімдіктер
Ақтөбеде сирек кездесетін өсімдіктер Систематика класса Птицы
Систематика класса Птицы 20231221_baranova_gmo
20231221_baranova_gmo Экология и природопользование. Экосистемы
Экология и природопользование. Экосистемы