- Секвенирование НК. Секвенирование по Сэнгеру. Занятие 7

Содержание
- 2. Технологические варианты секвенирования Методы секвенирования I поколения (по Сэнгеру, по Максаму – Гилберту, автоматическое секвенирование по
- 3. Методические варианты секвенирования 1. Секвенирование ДНК (геном) / Секвенирование РНК=>кДНК (транскриптом) 2. Ресеквенирование / Секвенирование de
- 4. Этапы секвенирования 1. Выделение ДНК. 2. Создание геномной библиотеки (полногеномное секвенирование) или амплификация участка ДНК (целевое
- 5. Геномные библиотеки (ДНК-библиотеки) Геномная библиотека – набор фрагментов одного генома, каждый фрагмент уникален. Для секвенирования необходимо
- 6. Клонирование в вектор
- 7. Создание ДНК-библиотек с помощью ПЦР
- 8. Концепция shotgun-секвенирования – получение геномных библиотек
- 9. Целевое (таргетное) секвенирование В процессе приготовления библиотеки необходимо выделить только целевые участки НК: 1). ПЦР со
- 10. Секвенирование по Максаму – Гилберту Основа метода – химическое расщепление множества копий одной целевой молекулы ДНК
- 11. Этапы секвенирования по Максаму – Гилберту 1). Получение множества копий целевой последовательности (100-1000 п.о.). 2). Мечение
- 12. Химический гидролиз по основаниям
- 13. Схема метода Максама – Гилберта
- 14. Характеристика секвенирования по Максаму – Гилберту Преимущества: Высокая точность Большая длина прочтения (300-1000 п.о.) Не требует
- 15. Основа метода Сэнгера: терминация синтеза [ddNTP]/[dNTP]=1/20
- 16. Классическая схема Сэнгера (1977 год)
- 17. Автоматическое секвенирование по Сэнгеру (1990 год)
- 18. Автоматический секвенатор (генетические анализатор) (Applied Biosystems 3500 xL)
- 19. Генетический анализатор с капиллярным электрофорезом изнутри
- 20. Капиллярный электрофорез
- 21. Лазерная детекция флюоресценции
- 22. Параметры метода (платформы) секвенирования Длина прочтения (длина рида, read length) Производительность (выход, output) Уровень ошибки Время
- 23. Секвенирование по Сэнгеру: Длина прочтения (длина рида) Результат секвенирования – набор последовательностей (прочтений) сходной длины. Длина
- 24. Секвенирование по Сэнгеру: Выход секвенирования (производительность) Выход равен количеству прочитанных нуклеотидов за один запуск: O=l*n l
- 25. Секвенирование по Сэнгеру: Количество образцов на один запуск Количество образцов определяется исходя из выхода секвенирования и
- 26. Секвенирование по Сэнгеру: Уровень ошибки секвенирования Уровень ошибки – доля нуклеотидов, прочитанных ошибочно (%). Источники ошибок
- 27. Секвенирование по Сэнгеру: Время одного запуска 2-3 часа: – 2 часа на терминирующую ПЦР – 40
- 28. Секвенирование по Сэнгеру: Стоимость (в пересчёте на производительность) Самый дорогой метод: стоимость выше ~ в 10
- 29. Характеристика секвенирования по Сэнгеру Преимущества: Низкая частота ошибок (0,1-1%) Большая длина прочтения (300-1000 п.о.) Недостатки: Низкая
- 31. Скачать презентацию

Технологические варианты секвенирования
Методы секвенирования I поколения (по Сэнгеру, по Максаму –
Технологические варианты секвенирования
Методы секвенирования I поколения (по Сэнгеру, по Максаму –

Методические варианты секвенирования
1. Секвенирование ДНК (геном) / Секвенирование РНК=>кДНК (транскриптом)
2. Ресеквенирование
Методические варианты секвенирования
1. Секвенирование ДНК (геном) / Секвенирование РНК=>кДНК (транскриптом)
2. Ресеквенирование

Этапы секвенирования
1. Выделение ДНК.
2. Создание геномной библиотеки (полногеномное секвенирование) или амплификация
Этапы секвенирования
1. Выделение ДНК.
2. Создание геномной библиотеки (полногеномное секвенирование) или амплификация

Геномные библиотеки (ДНК-библиотеки)
Геномная библиотека – набор фрагментов одного генома, каждый фрагмент
Геномные библиотеки (ДНК-библиотеки)
Геномная библиотека – набор фрагментов одного генома, каждый фрагмент
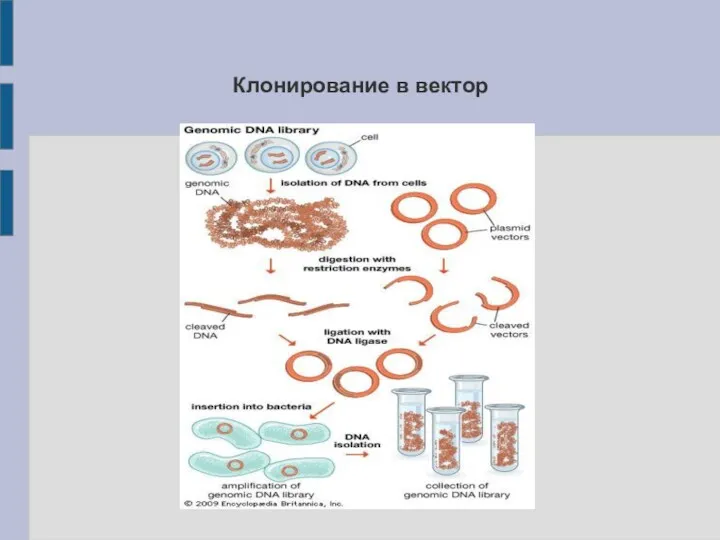
Клонирование в вектор
Клонирование в вектор

Создание ДНК-библиотек с помощью ПЦР
Создание ДНК-библиотек с помощью ПЦР
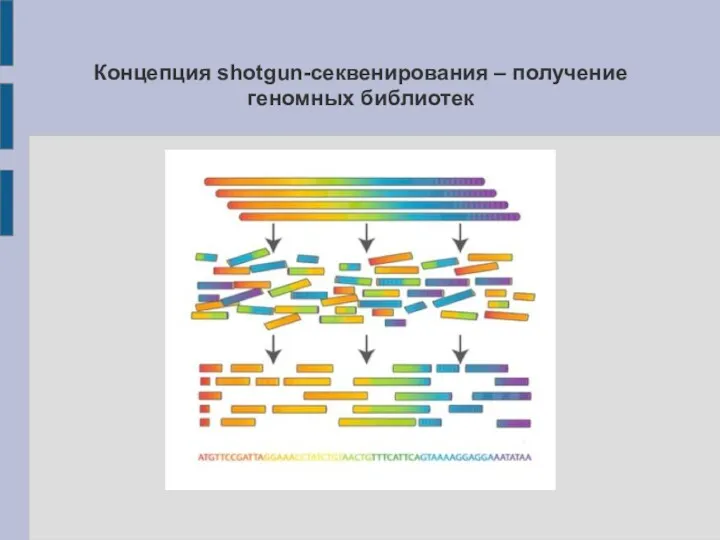
Концепция shotgun-секвенирования – получение геномных библиотек
Концепция shotgun-секвенирования – получение геномных библиотек

Целевое (таргетное) секвенирование
В процессе приготовления библиотеки необходимо выделить только целевые участки
Целевое (таргетное) секвенирование
В процессе приготовления библиотеки необходимо выделить только целевые участки

Секвенирование по Максаму – Гилберту
Основа метода – химическое расщепление множества копий
Секвенирование по Максаму – Гилберту
Основа метода – химическое расщепление множества копий

Этапы секвенирования по Максаму – Гилберту
1). Получение множества копий целевой последовательности
Этапы секвенирования по Максаму – Гилберту
1). Получение множества копий целевой последовательности
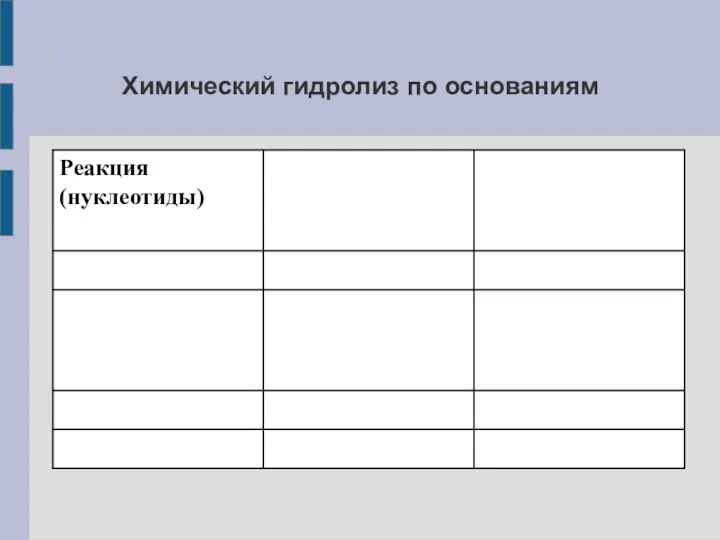
Химический гидролиз по основаниям
Химический гидролиз по основаниям
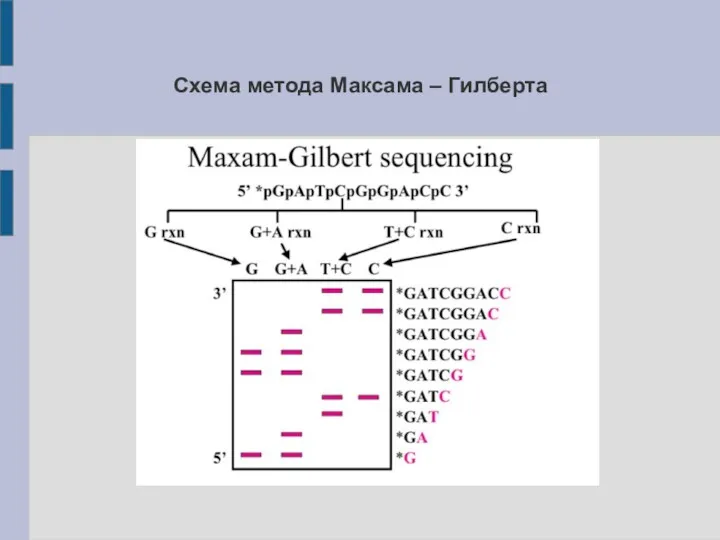
Схема метода Максама – Гилберта
Схема метода Максама – Гилберта

Характеристика секвенирования по Максаму – Гилберту
Преимущества:
Высокая точность
Большая длина прочтения (300-1000 п.о.)
Не
Характеристика секвенирования по Максаму – Гилберту
Преимущества:
Высокая точность
Большая длина прочтения (300-1000 п.о.)
Не
![Основа метода Сэнгера: терминация синтеза [ddNTP]/[dNTP]=1/20](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/232827/slide-14.jpg)
Основа метода Сэнгера: терминация синтеза
[ddNTP]/[dNTP]=1/20
Основа метода Сэнгера: терминация синтеза
[ddNTP]/[dNTP]=1/20

Классическая схема Сэнгера (1977 год)
Классическая схема Сэнгера (1977 год)

Автоматическое секвенирование по Сэнгеру (1990 год)
Автоматическое секвенирование по Сэнгеру (1990 год)

Автоматический секвенатор (генетические анализатор) (Applied Biosystems 3500 xL)
Автоматический секвенатор (генетические анализатор) (Applied Biosystems 3500 xL)

Генетический анализатор с капиллярным электрофорезом изнутри
Генетический анализатор с капиллярным электрофорезом изнутри

Капиллярный электрофорез
Капиллярный электрофорез

Лазерная детекция флюоресценции
Лазерная детекция флюоресценции

Параметры метода (платформы) секвенирования
Длина прочтения (длина рида, read length)
Производительность (выход, output)
Уровень
Параметры метода (платформы) секвенирования
Длина прочтения (длина рида, read length)
Производительность (выход, output)
Уровень
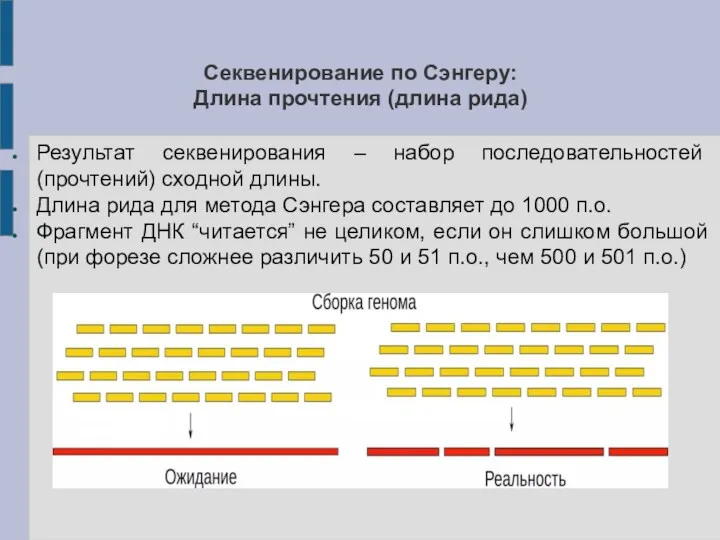
Секвенирование по Сэнгеру:
Длина прочтения (длина рида)
Результат секвенирования – набор последовательностей (прочтений)
Секвенирование по Сэнгеру:
Длина прочтения (длина рида)
Результат секвенирования – набор последовательностей (прочтений)

Секвенирование по Сэнгеру:
Выход секвенирования (производительность)
Выход равен количеству прочитанных нуклеотидов за один
Секвенирование по Сэнгеру:
Выход секвенирования (производительность)
Выход равен количеству прочитанных нуклеотидов за один

Секвенирование по Сэнгеру:
Количество образцов на один запуск
Количество образцов определяется исходя из
Секвенирование по Сэнгеру:
Количество образцов на один запуск
Количество образцов определяется исходя из

Секвенирование по Сэнгеру:
Уровень ошибки секвенирования
Уровень ошибки – доля нуклеотидов, прочитанных ошибочно
Секвенирование по Сэнгеру:
Уровень ошибки секвенирования
Уровень ошибки – доля нуклеотидов, прочитанных ошибочно

Секвенирование по Сэнгеру:
Время одного запуска
2-3 часа:
– 2 часа на терминирующую ПЦР
–
Секвенирование по Сэнгеру:
Время одного запуска
2-3 часа:
– 2 часа на терминирующую ПЦР
–

Секвенирование по Сэнгеру:
Стоимость (в пересчёте на производительность)
Самый дорогой метод: стоимость выше
Секвенирование по Сэнгеру:
Стоимость (в пересчёте на производительность)
Самый дорогой метод: стоимость выше

Характеристика секвенирования по Сэнгеру
Преимущества:
Низкая частота ошибок (0,1-1%)
Большая длина прочтения (300-1000 п.о.)
Недостатки:
Низкая
Характеристика секвенирования по Сэнгеру
Преимущества:
Низкая частота ошибок (0,1-1%)
Большая длина прочтения (300-1000 п.о.)
Недостатки:
Низкая
 Нуклеїнові кислоти (ДНК та РНК)
Нуклеїнові кислоти (ДНК та РНК) Botanika. Organizace předmětu
Botanika. Organizace předmětu Генетика пола
Генетика пола Биологиялық мембраналардың өткізгіштік механизмі. Иондық каналдардың және тасымалдаушылардың құрылысы мен функциясы
Биологиялық мембраналардың өткізгіштік механизмі. Иондық каналдардың және тасымалдаушылардың құрылысы мен функциясы Клонування організмів
Клонування організмів Урок – практикум. Генеалогический метод исследования живых организмов. Составление родословной А. С. Пушкина
Урок – практикум. Генеалогический метод исследования живых организмов. Составление родословной А. С. Пушкина Молекулярные механизмы образования хромосомных перестроек с учетом структурной организации хромосомных районов
Молекулярные механизмы образования хромосомных перестроек с учетом структурной организации хромосомных районов Поширення плодів та насіння у природі
Поширення плодів та насіння у природі Конспект урока: Экологические типы птиц, их роль в природе, жизни человека
Конспект урока: Экологические типы птиц, их роль в природе, жизни человека Імунна система людини, особливості її функціонування
Імунна система людини, особливості її функціонування Кайнозойская эра. Антропогеновый период
Кайнозойская эра. Антропогеновый период Применение активных дрожжей в кормлений животных
Применение активных дрожжей в кормлений животных Клетка: строение и функции (цитология)
Клетка: строение и функции (цитология) Виды регуляций в организме
Виды регуляций в организме Мочеполовой аппарат
Мочеполовой аппарат Ядовитые растения Курганской области
Ядовитые растения Курганской области Почему лук щиплет глаза?
Почему лук щиплет глаза? Цветок. Строение цветка
Цветок. Строение цветка Плазуни. Зовнішня будова
Плазуни. Зовнішня будова Почему человек потеет
Почему человек потеет презентация к уроку Пищеварение в кишечнике
презентация к уроку Пищеварение в кишечнике Профилактика сердечно-сосудистых заболеваний
Профилактика сердечно-сосудистых заболеваний Обмен веществ и энергии, терморегуляция
Обмен веществ и энергии, терморегуляция Условные и безусловные рефлексы
Условные и безусловные рефлексы Обоняние, осязание, вкус
Обоняние, осязание, вкус Fruits and their classification. Spreading of fruits and seeds
Fruits and their classification. Spreading of fruits and seeds Функциональная анатомия женских половых органов
Функциональная анатомия женских половых органов Эпифиз (пинеальная железа, шишковидное тело, corpus pineale, epiphysis cerebri)
Эпифиз (пинеальная железа, шишковидное тело, corpus pineale, epiphysis cerebri)